Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders

 , , and
, , and
Abstract
:1. Neuroinflammation in Neurodegenerative Disorders
1.1. Neuroinflammation Overview
1.2. Neuroinflammation in Neurodegenerative Diseases
1.3. Monitoring Neuroinflammation as A Biomarker in Neurodegenerative Diseases
2. TSPO as a Biomarker For Neuroinflammation
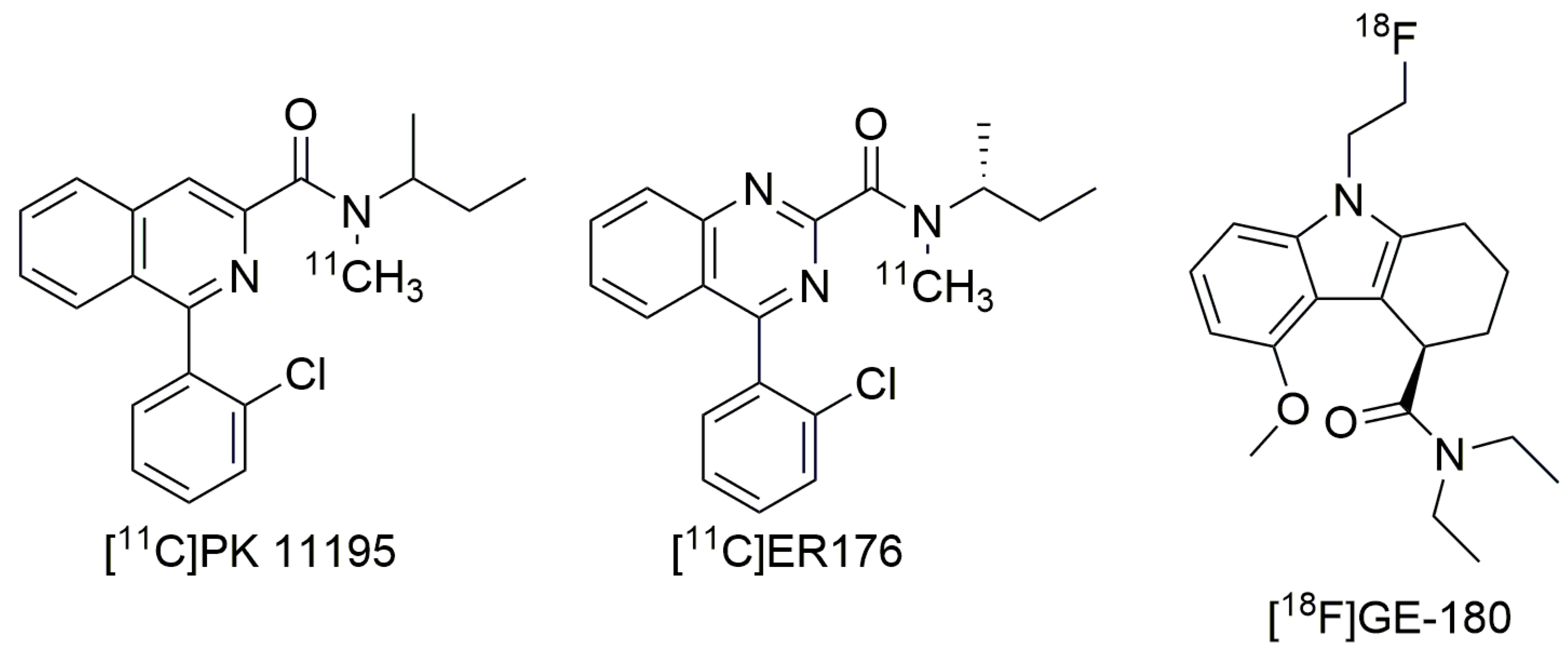
3. Third-Generation Ligands to Overcome The Challenge of A147T TSPO
3.1. ER176 and GE-180
3.2. Future Directions
4. Cellular and Functional Interpretation of TSPO PET Signals
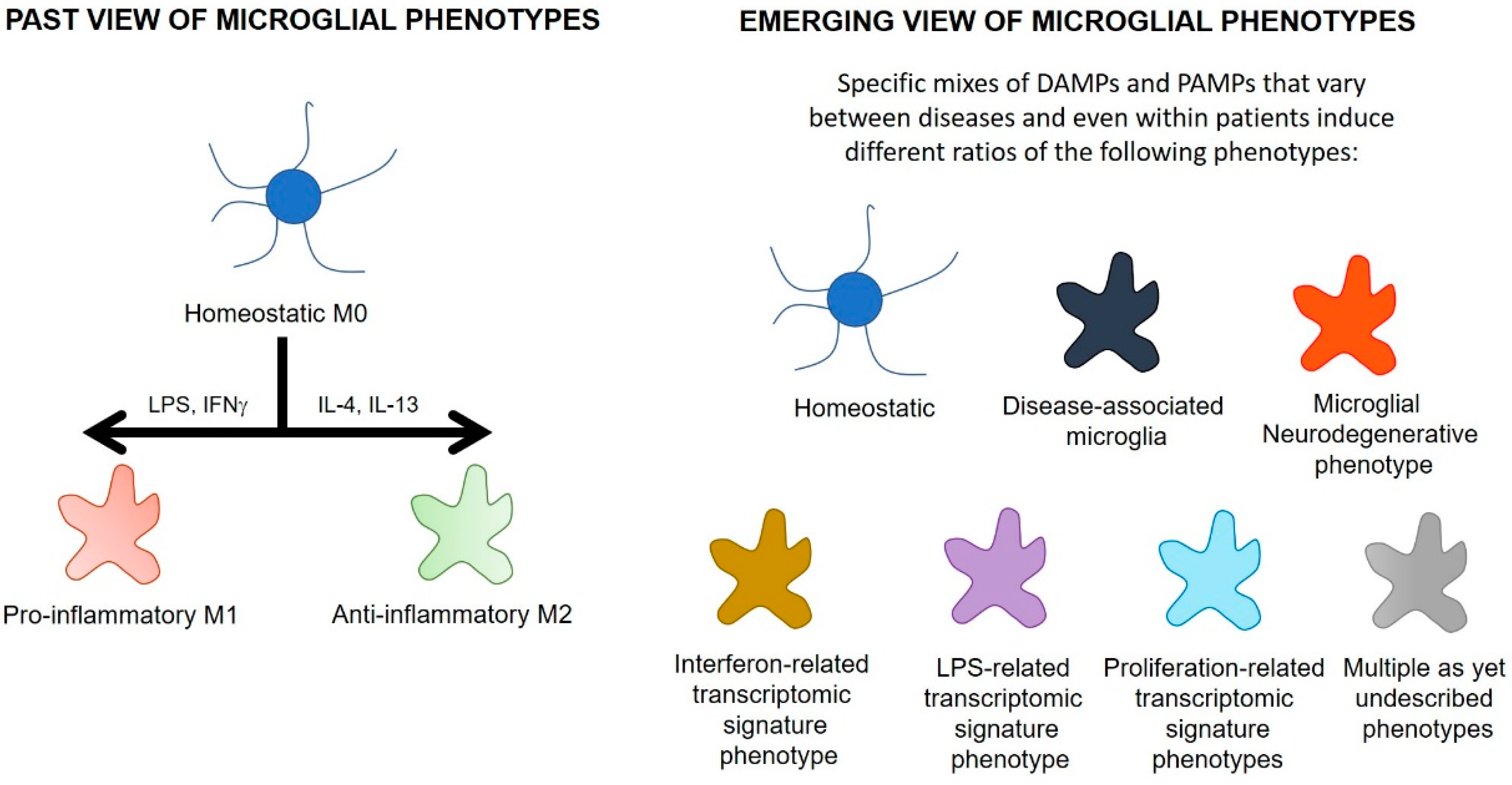
4.1. Microglial Phenotypes
4.2. Astrocytes
4.3. Neurons
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| A147T TSPO | Ala147Thr translocator protein |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| BBB | blood-brain barrier |
| BPND | binding potential |
| CNS | central nervous system |
| DAMPs | danger-associated molecular patterns |
| FTD | frontotemporal dementia |
| GFAP | glial fibrillary acidic protein |
| GWAS | genome-wide association studies |
| HAB | high affinity binder |
| HLA | human leukocyte antigen |
| IFNγ | interferon-γ |
| IL | interleukin |
| Ki | affinity |
| LAB | low affinity binder |
| LPS | lipopolysaccharide |
| MAB | mixed affinity binder |
| PAMPs | pathogen-associated molecular patterns |
| PET | positron emission tomography |
| MAB | mixed affinity binder |
| PAMPs | pathogen-associated molecular patterns |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| PRRs | pattern recognition receptors |
| RsTSPO | Rhodobacter sphaeroides translocator protein |
| SNP | single nucleotide polymorphism |
| TLRs | toll-like receptors |
| TSPO | translocator protein |
| WT TSPO | wild type translocator protein |
References
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzeriat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1. [Google Scholar]
- O’Callaghan, J.P.; Sriram, K.; Miller, D.B. Defining “neuroinflammation”. Ann. N. Y. Acad. Sci. 2008, 1139, 318–330. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.; Kaat, L.D.; Rohrer, J.D.; van Swieten, J.C. Imaging and fluid biomarkers in frontotemporal dementia. Nat. Rev. Neurol. 2017, 13, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Gratwicke, J.; Jahanshahi, M.; Foltynie, T. Parkinson’s disease dementia: A neural networks perspective. Brain J. Neurol. 2015, 138, 1454–1476. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Hernandez, D.G.; Nalls, M.A.; Rohrer, J.D.; Ramasamy, A.; Kwok, J.B.; Dobson-Stone, C.; Brooks, W.S.; Schofield, P.R.; Halliday, G.M.; et al. Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurol. 2014, 13, 686–699. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Consortium, U.K.B.E.; North American Brain Expression, C.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bevan-Jones, W.R.; Cope, T.E.; Jones, P.S.; Passamonti, L.; Hong, Y.T.; Fryer, T.; Arnold, R.; Coles, J.P.; Aigbirhio, F.I.; O’Brien, J.T.; et al. In vivo evidence for pre-symptomatic neuroinflammation in a MAPT mutation carrier. Ann. Clin. Transl. Neurol. 2019, 6, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef]
- Cebrian, C.; Loike, J.D.; Sulzer, D. Neuroinflammation in Parkinson’s disease animal models: A cell stress response or a step in neurodegeneration? Curr. Top. Behav. Neurosci. 2015, 22, 237–270. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Paterson, R.W.; Warren, J.D.; Zetterberg, H.; O’Brien, J.T.; Fox, N.C.; Halliday, G.M.; Schott, J.M. Biomarkers in dementia: Clinical utility and new directions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1426–1434. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth Factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.M.; Ke, Y.D.; Vucic, S.; Ittner, L.M.; Seeley, W.; Hodges, J.R.; Piguet, O.; Halliday, G.; Kiernan, M.C. Physiological changes in neurodegeneration—Mechanistic insights and clinical utility. Nat. Rev. Neurol. 2018, 14, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.; da Cruz, E.S.O.A.B.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, B.; Vas, A.; Toth, M.; Takano, A.; Varrone, A.; Cselenyi, Z.; Schain, M.; Mattsson, P.; Halldin, C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with [11C]vinpocetine. Neuroimage 2011, 56, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Chung, J.H.; Choi, T.K.; Suh, S.Y.; Oh, B.H.; Hong, C.H. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Venturelli, E.; Fenoglio, C.; Guidi, I.; Villa, C.; Bergamaschini, L.; Cortini, F.; Scalabrini, D.; Baron, P.; Vergani, C.; et al. Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degeneration. J. Neurol. 2008, 255, 539–544. [Google Scholar] [CrossRef]
- Sjogren, M.; Folkesson, S.; Blennow, K.; Tarkowski, E. Increased intrathecal inflammatory activity in frontotemporal dementia: Pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1107–1111. [Google Scholar] [CrossRef]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009381. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Cook, D.A.; Lee, J.K.; Chang, J.; Chung, J.; Sandy, E.; Paul, K.C.; Ritz, B.; Bronstein, J.; Factor, S.A.; et al. Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson’s Disease: An Observational and Case-Control Study. NPJ Parkinsons Dis. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Neuroinflammation and ALS: Transcriptomic Insights into Molecular Disease Mechanisms and Therapeutic Targets. Mediat. Inflamm. 2017, 2017, 7070469. [Google Scholar] [CrossRef] [PubMed]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Hoglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Gagliano, S.A.; Pouget, J.G.; Hardy, J.; Knight, J.; Barnes, M.R.; Ryten, M.; Weale, M.E. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann. Clin. Transl. Neurol. 2016, 3, 924–933. [Google Scholar] [CrossRef]
- Fogh, I.; Ratti, A.; Gellera, C.; Lin, K.; Tiloca, C.; Moskvina, V.; Corrado, L.; Soraru, G.; Cereda, C.; Corti, S.; et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 2220–2231. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef]
- Mora, J.S.; Barbeito, L.; Hermine, O. Masitinib as an Add-On Therapy to Riluzole Is Beneficial in the Treatment of Amyotrophic Lateral Sclerosis (ALS) with Acceptable Tolerability: Results from a Randomized Controlled Phase 3 Trial. European Network to Cure ALS (ENCALS). 2017. Available online: http://videolectures.net/encals2017_barbeito_mora_hermine_therapy/ (accessed on 19 May 2019).
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Forum on Neuroscience and Nervous System Disorders. Biomarkers of Neuroinflammation: Proceedings of a Workshop; National Academies Press: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Politis, M.; Su, P.; Piccini, P. Imaging of microglia in patients with neurodegenerative disorders. Front. Pharmacol. 2012, 3, 96. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Loth, M.K.; Guariglia, S.R. TSPO Finds NOX2 in Microglia for Redox Homeostasis. Trends Pharmacol. Sci. 2016, 37, 334–343. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Campioli, E.; Midzak, A.; Culty, M.; Papadopoulos, V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc. Natl. Acad. Sci. USA 2015, 112, 7261–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, V.; Tu, L.N.; Stocco, D.M. Crucial Role Reported for TSPO in Viability and Steroidogenesis is a Misconception. Commentary: Conditional Steroidogenic Cell-Targeted Deletion of TSPO Unveils a Crucial Role in Viability and Hormone-Dependent Steroid Formation. Front. Endocrinol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Martini, C. Translocator protein and steroidogenesis. Biochem. J. 2018, 475, 901–904. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [Green Version]
- Abourbeh, G.; Theze, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dolle, F.; Tavitian, B.; Boisgard, R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef]
- Amhaoul, H.; Hamaide, J.; Bertoglio, D.; Reichel, S.N.; Verhaeghe, J.; Geerts, E.; Van Dam, D.; De Deyn, P.P.; Kumar-Singh, S.; Katsifis, A.; et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol. Dis. 2015, 82, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.L.; Beck, R.; Lindner, S.; Gildehaus, F.J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, D.J.; Selvaraj, V.; Chechneva, O.V.; Liu, X.B.; Pleasure, D.E.; Deng, W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 2013, 5, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerdere, S.; Callaghan, P.D.; Pham, T.; Rahardjo, G.L.; Amhaoul, H.; Berghofer, P.; Quinlivan, M.; Mattner, F.; Loc’h, C.; Katsifis, A.; et al. PET imaging of brain inflammation during early epileptogenesis in a rat model of temporal lobe epilepsy. EJNMMI Res. 2012, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Israel, I.; Ohsiek, A.; Al-Momani, E.; Albert-Weissenberger, C.; Stetter, C.; Mencl, S.; Buck, A.K.; Kleinschnitz, C.; Samnick, S.; Siren, A.L. Combined [18F]DPA-714 micro-positron emission tomography and autoradiography imaging of microglia activation after closed head injury in mice. J. Neuroinflamm. 2016, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Boisgard, R.; Theze, B.; Van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dolle, F.; Tavitian, B. Evaluation of the PBR/TSPO radioligand [18F]DPA-714 in a rat model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Katsifis, A.; Staykova, M.; Ballantyne, P.; Willenborg, D.O. Evaluation of a radiolabelled peripheral benzodiazepine receptor ligand in the central nervous system inflammation of experimental autoimmune encephalomyelitis: A possible probe for imaging multiple sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Tang, S.P.; Ashworth, S.; Coello, C.; Plisson, C.; Passchier, J.; Selvaraj, V.; Tyacke, R.J.; Nutt, D.J.; Sastre, M. In vivo imaging of microglial activation by positron emission tomography with [11C]PBR28 in the 5XFAD model of Alzheimer’s disease. Glia 2016, 64, 993–1006. [Google Scholar] [CrossRef]
- Thomas, C.; Vercouillie, J.; Domene, A.; Tauber, C.; Kassiou, M.; Guilloteau, D.; Destrieux, C.; Serriere, S.; Chalon, S. Detection of Neuroinflammation in a Rat Model of Subarachnoid Hemorrhage Using [18F]DPA-714 PET Imaging. Mol. Imaging 2016, 15. [Google Scholar] [CrossRef]
- Toth, M.; Little, P.; Arnberg, F.; Haggkvist, J.; Mulder, J.; Halldin, C.; Gulyas, B.; Holmin, S. Acute neuroinflammation in a clinically relevant focal cortical ischemic stroke model in rat: Longitudinal positron emission tomography and immunofluorescent tracking. Brain Struct. Funct. 2016, 221, 1279–1290. [Google Scholar] [CrossRef]
- Tremoleda, J.L.; Thau-Zuchman, O.; Davies, M.; Foster, J.; Khan, I.; Vadivelu, K.C.; Yip, P.K.; Sosabowski, J.; Trigg, W.; Michael-Titus, A.T. In vivo PET imaging of the neuroinflammatory response in rat spinal cord injury using the TSPO tracer [18F]GE-180 and effect of docosahexaenoic acid. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1710–1722. [Google Scholar] [CrossRef]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: Preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Belichenko, N.P.; Shuhendler, A.J.; Hoehne, A.; Andrews, L.E.; Condon, C.; Nguyen, T.V.; Reiser, V.; Jones, P.; Trigg, W.; et al. [18F]GE-180 PET Detects Reduced Microglia Activation After LM11A-31 Therapy in a Mouse Model of Alzheimer’s Disease. Theranostics 2017, 7, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, B.; Toth, M.; Schain, M.; Airaksinen, A.; Vas, A.; Kostulas, K.; Lindstrom, P.; Hillert, J.; Halldin, C. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: A PET study with the TSPO molecular imaging biomarker [11C]vinpocetine. J. Neurol. Sci. 2012, 320, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Takano, A.; Piehl, F.; Hillert, J.; Varrone, A.; Nag, S.; Gulyas, B.; Stenkrona, P.; Villemagne, V.L.; Rowe, C.C.; Macdonell, R.; et al. In vivo TSPO imaging in patients with multiple sclerosis: A brain PET study with [18F]FEDAA1106. EJNMMI Res. 2013, 3, 30. [Google Scholar] [CrossRef]
- Golla, S.S.; Boellaard, R.; Oikonen, V.; Hoffmann, A.; van Berckel, B.N.; Windhorst, A.D.; Virta, J.; Haaparanta-Solin, M.; Luoto, P.; Savisto, N.; et al. Quantification of [18F]DPA-714 binding in the human brain: Initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015, 35, 766–772. [Google Scholar] [CrossRef]
- Varrone, A.; Mattsson, P.; Forsberg, A.; Takano, A.; Nag, S.; Gulyas, B.; Borg, J.; Boellaard, R.; Al-Tawil, N.; Eriksdotter, M.; et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, F.; Van Camp, N.; Dolle, F.; Kuhnast, B.; Hinnen, F.; Damont, A.; Boutin, H.; James, M.; Kassiou, M.; Tavitian, B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J. Nucl. Med. 2009, 50, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Howell, O.W.; Tang, S.P.; Wells, L.A.; Bennacef, I.; Bergstrom, M.; Gunn, R.N.; Rabiner, E.A.; Wilkins, M.R.; Reynolds, R.; et al. Two binding sites for [3H]PBR28 in human brain: Implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2010, 30, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef]
- Mizrahi, R.; Rusjan, P.M.; Kennedy, J.; Pollock, B.; Mulsant, B.; Suridjan, I.; De Luca, V.; Wilson, A.A.; Houle, S. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J. Cereb. Blood Flow Metab. 2012, 32, 968–972. [Google Scholar] [CrossRef]
- Owen, D.R.; Lewis, A.J.; Reynolds, R.; Rupprecht, R.; Eser, D.; Wilkins, M.R.; Bennacef, I.; Nutt, D.J.; Parker, C.A. Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocator protein in human brain. Synapse 2011, 65, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Colasanti, A.; Owen, D.R.; Onega, M.; Kamalakaran, A.; Bennacef, I.; Matthews, P.M.; Rabiner, E.A.; Turkheimer, F.E.; Gunn, R.N. Quantification of the specific translocator protein signal of 18F-PBR111 in healthy humans: A genetic polymorphism effect on in vivo binding. J. Nucl. Med. 2013, 54, 1915–1923. [Google Scholar] [CrossRef]
- Colasanti, A.; Guo, Q.; Muhlert, N.; Giannetti, P.; Onega, M.; Newbould, R.D.; Ciccarelli, O.; Rison, S.; Thomas, C.; Nicholas, R.; et al. In Vivo Assessment of Brain White Matter Inflammation in Multiple Sclerosis with 18F-PBR111 PET. J. Nucl. Med. 2014, 55, 1112–1118. [Google Scholar] [CrossRef]
- Zurcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E.; et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28. Neuroimage Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Suridjan, I.; Pollock, B.G.; Verhoeff, N.P.; Voineskos, A.N.; Chow, T.; Rusjan, P.M.; Lobaugh, N.J.; Houle, S.; Mulsant, B.H.; Mizrahi, R. In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: A positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol. Psychiatry 2015, 20, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. 11C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Colasanti, A.; Kalk, N.; Owen, D.; Scott, G.; Rabiner, E.A.; Gunn, R.N.; Lingford-Hughes, A.; Malik, O.; Ciccarelli, O.; et al. 11C-PBR28 and 18F-PBR111 Detect White Matter Inflammatory Heterogeneity in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1477–1482. [Google Scholar] [CrossRef]
- Varrone, A.; Oikonen, V.; Forsberg, A.; Joutsa, J.; Takano, A.; Solin, O.; Haaparanta-Solin, M.; Nag, S.; Nakao, R.; Al-Tawil, N.; et al. Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer’s disease patients and control subjects. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 438–446. [Google Scholar] [CrossRef]
- Lyoo, C.H.; Ikawa, M.; Liow, J.S.; Zoghbi, S.S.; Morse, C.L.; Pike, V.W.; Fujita, M.; Innis, R.B.; Kreisl, W.C. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J. Nucl. Med. 2015, 56, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Yoder, K.K.; Nho, K.; Risacher, S.L.; Kim, S.; Shen, L.; Saykin, A.J. Influence of TSPO genotype on 11C-PBR28 standardized uptake values. J. Nucl. Med. 2013, 54, 1320–1322. [Google Scholar] [CrossRef]
- Sucksdorff, M.; Rissanen, E.; Tuisku, J.; Nuutinen, S.; Paavilainen, T.; Rokka, J.; Rinne, J.; Airas, L. Evaluation of the Effect of Fingolimod Treatment on Microglial Activation Using Serial PET Imaging in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1646–1651. [Google Scholar] [CrossRef] [Green Version]
- Chauveau, F.; Boutin, H.; Van Camp, N.; Dolle, F.; Tavitian, B. Nuclear imaging of neuroinflammation: A comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Zhang, Y.; Jenko, K.J.; Gladding, R.L.; Zoghbi, S.S.; Fujita, M.; Sbardella, G.; Castellano, S.; Taliani, S.; Martini, C.; et al. Synthesis and evaluation of translocator 18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci. 2014, 5, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kobayashi, M.; Ikawa, M.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Zoghbi, S.S.; Haskali, M.B.; Telu, S.; Pike, V.W.; et al. Comparison of four 11C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Lohith, T.G.; Shrestha, S.; Telu, S.; Zoghbi, S.S.; Castellano, S.; Taliani, S.; Da Settimo, F.; Fujita, M.; Pike, V.W.; et al. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J. Nucl. Med. 2017, 58, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Stoll, H.P.; Hutchins, G.D.; Winkle, W.L.; Nguyen, A.T.; Appledorn, C.R.; Janzen, I.; Seifert, H.; Rube, C.; Schieffer, H.; March, K.L. Advantages of short-lived positron-emitting radioisotopes for intracoronary radiation therapy with liquid-filled balloons to prevent restenosis. J. Nucl. Med. 2001, 42, 1375–1383. [Google Scholar] [PubMed]
- Liu, B.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Holton, P.; Reiser, V.; Jones, P.A.; et al. In Vivo Detection of Age- and Disease-Related Increases in Neuroinflammation by 18F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer’s Transgenic Mice. J. Neurosci. 2015, 35, 15716–15730. [Google Scholar] [CrossRef]
- Boutin, H.; Murray, K.; Pradillo, J.; Maroy, R.; Smigova, A.; Gerhard, A.; Jones, P.A.; Trigg, W. 18F-GE-180: A novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Picon, F.R.; Snellman, A.; Eskola, O.; Helin, S.; Solin, O.; Haaparanta-Solin, M.; Rinne, J.O. Neuroinflammation Appears Early on PET Imaging and Then Plateaus in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2018, 59, 509–515. [Google Scholar] [CrossRef]
- Sridharan, S.; Lepelletier, F.X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. 11C-DPA-713 Versus 18F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef]
- Unterrainer, M.; Mahler, C.; Vomacka, L.; Lindner, S.; Havla, J.; Brendel, M.; Boning, G.; Ertl-Wagner, B.; Kumpfel, T.; Milenkovic, V.M.; et al. TSPO PET with [18F]GE-180 sensitively detects focal neuroinflammation in patients with relapsing-remitting multiple sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Fisher, E.; McMurray, L.; Milicevic Sephton, S.; Hird, M.; Kuzhuppilly-Ramakrishnan, N.; Williamson, D.J.; Zhou, X.; Werry, E.; Kassiou, M.; et al. Radiosynthesis of (R,S)-[18F]GE387: A Potential PET Radiotracer for Imaging Translocator Protein 18 kDa (TSPO) with Low Binding Sensitivity to the Human Gene Polymorphism rs6971. ChemMedChem 2019, 14, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Raffel, J.; Nandoskar, A.; Record, C.; Brooks, D.J.; Owen, D.; Sharp, D.; Muraro, P.A.; Gunn, R.; Nicholas, R. Confirmation of Specific Binding of the 18-kDa Translocator Protein (TSPO) Radioligand [18F]GE-180: A Blocking Study Using XBD173 in Multiple Sclerosis Normal Appearing White and Grey Matter. Mol. Imaging Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zanotti-Fregonara, P.; Veronese, M.; Pascual, B.; Rostomily, R.C.; Turkheimer, F.; Masdeu, J.C. The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.L.; Unterrainer, M.; Brendel, M.; Kaiser, L.; Zweckstetter, M.; Cumming, P.; Bartenstein, P. In response to: The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, M.; Jaremko, L.; Giller, K.; Becker, S.; Zweckstetter, M. Backbone and side-chain resonance assignment of the A147T polymorph of mouse TSPO in complex with a high-affinity radioligand. Biomol. NMR Assign 2016, 10, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, M.; Jaremko, L.; Giller, K.; Becker, S.; Zweckstetter, M. Structural Integrity of the A147T Polymorph of Mammalian TSPO. Chembiochem 2015, 16, 1483–1489. [Google Scholar] [CrossRef]
- Berroteran-Infante, N.; Tadic, M.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Binding Affinity of Some Endogenous and Synthetic TSPO Ligands Regarding the rs6971 Polymorphism. Int. J. Mol. Sci. 2019, 20, 563. [Google Scholar] [CrossRef]
- Sokias, R.; Werry, E.L.; Chua, S.W.; Reekie, T.A.; Munoz, L.; Wong, E.C.N.; Ittner, L.M.; Kassiou, M. Determination and reduction of translocator protein (TSPO) ligand rs6971 discrimination. Medchemcomm 2017, 8, 202–210. [Google Scholar] [CrossRef]
- Midzak, A.S.; Akula, N.; Rone, M.B.; Papadopoulos, V. Computational modeling and biological validation of novel non-steroidal ligands for the cholesterol recognition/interaction amino acid consensus (CRAC) motif of the mitochondrial translocator protein (TSPO). Pharmacol. Res. 2015, 99, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rone, M.B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol. 2012, 26, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Stathis, M.; Coughlin, J.M.; Pomper, M.; Slusher, B.S. The Low-Affinity Binding of Second Generation Radiotracers Targeting TSPO is Associated with a Unique Allosteric Binding Site. J. Neuroimmune Pharmacol. 2018, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Schober, D.A.; Smalstig, E.B.; Mincy, R.E.; Gehlert, D.R.; Clemens, J.A. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J. Neurosci. 1995, 15, 5263–5274. [Google Scholar] [CrossRef] [PubMed]
- Vowinckel, E.; Reutens, D.; Becher, B.; Verge, G.; Evans, A.; Owens, T.; Antel, J.P. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1997, 50, 345–353. [Google Scholar] [CrossRef]
- Arlicot, N.; Katsifis, A.; Garreau, L.; Mattner, F.; Vergote, J.; Duval, S.; Kousignian, I.; Bodard, S.; Guilloteau, D.; Chalon, S. Evaluation of CLINDE as potent translocator protein (18 kDa) SPECT radiotracer reflecting the degree of neuroinflammation in a rat model of microglial activation. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Baidoo, K.; Verina, T.; Guilarte, T.R. Peripheral benzodiazepine receptor imaging in CNS demyelination: Functional implications of anatomical and cellular localization. Brain 2004, 127, 1379–1392. [Google Scholar] [CrossRef]
- Chen, M.K.; Guilarte, T.R. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol. Sci. 2006, 91, 532–539. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Beckers, L.; Ory, D.; Geric, I.; Declercq, L.; Koole, M.; Kassiou, M.; Bormans, G.; Baes, M. Increased Expression of Translocator Protein (TSPO) Marks Pro-inflammatory Microglia but Does Not Predict Neurodegeneration. Mol. Imaging Biol. 2018, 20, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustenhoven, J.; Park, T.I.; Schweder, P.; Scotter, J.; Correia, J.; Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; et al. Isolation of highly enriched primary human microglia for functional studies. Sci. Rep. 2016, 6, 19371. [Google Scholar] [CrossRef] [PubMed]
- Sandiego, C.M.; Gallezot, J.D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.F.; Matuskey, D.; Lee, J.Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef] [Green Version]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef]
- Chamberlain, L.M.; Holt-Casper, D.; Gonzalez-Juarrero, M.; Grainger, D.W. Extended culture of macrophages from different sources and maturation results in a common M2 phenotype. J. Biomed. Mater. Res. A 2015, 103, 2864–2874. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.L.; Gundlach, A.L.; Craven, J.A. Temporal changes in glial fibrillary acidic protein messenger RNA and [3H]PK11195 binding in relation to imidazoline-I2-receptor and alpha 2-adrenoceptor binding in the hippocampus following transient global forebrain ischaemia in the rat. Neuroscience 1998, 82, 805–817. [Google Scholar] [CrossRef]
- Myers, R.; Manjil, L.G.; Cullen, B.M.; Price, G.W.; Frackowiak, R.S.; Cremer, J.E. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J. Cereb. Blood Flow Metab. 1991, 11, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Domene, A.; Cavanagh, C.; Page, G.; Bodard, S.; Klein, C.; Delarasse, C.; Chalon, S.; Krantic, S. Expression of Phenotypic Astrocyte Marker Is Increased in a Transgenic Mouse Model of Alzheimer’s Disease versus Age-Matched Controls: A Presymptomatic Stage Study. Int. J. Alzheimers Dis. 2016, 2016, 5696241. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Kuhlmann, A.C.; O’Callaghan, J.P.; Miceli, R.C. Enhanced expression of peripheral benzodiazepine receptors in trimethyltin-exposed rat brain: A biomarker of neurotoxicity. Neurotoxicology 1995, 16, 441–450. [Google Scholar] [PubMed]
- Lavisse, S.; Guillermier, M.; Herard, A.S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dolle, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef]
- Karchewski, L.A.; Bloechlinger, S.; Woolf, C.J. Axonal injury-dependent induction of the peripheral benzodiazepine receptor in small-diameter adult rat primary sensory neurons. Eur. J. Neurosci. 2004, 20, 671–683. [Google Scholar] [CrossRef]
- Varga, B.; Marko, K.; Hadinger, N.; Jelitai, M.; Demeter, K.; Tihanyi, K.; Vas, A.; Madarasz, E. Translocator protein (TSPO 18kDa) is expressed by neural stem and neuronal precursor cells. Neurosci. Lett. 2009, 462, 257–262. [Google Scholar] [CrossRef]
- Bonsack, F.t.; Alleyne, C.H., Jr.; Sukumari-Ramesh, S. Augmented expression of TSPO after intracerebral hemorrhage: A role in inflammation? J. Neuroinflamm. 2016, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Mages, K.; Grassmann, F.; Jagle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflamm. 2019, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Notter, T.; Coughlin, J.M.; Gschwind, T.; Weber-Stadlbauer, U.; Wang, Y.; Kassiou, M.; Vernon, A.C.; Benke, D.; Pomper, M.G.; Sawa, A.; et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol. Psychiatry 2018, 23, 323–334. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Microglial Phenotype | Disease Model | Species | Features | Reference |
|---|---|---|---|---|
| Disease-associated microglia | Alzheimer’s disease | Mouse, human | Downregulated homeostatic genes (inc P2YR12, P2YR13, Tmem119, CX3CR1) and upregulated lysosomal and lipid metabolism-related genes (inc Apoe, Ctsd, Lpl, Tyrobp, TREM2). TSPO is upregulated 2.5x in disease-associated microglia. | [142,152] |
| Pro-inflammatory disease-associated microglia | Alzheimer’s disease | Mouse | Emerge earlier in the disease. Characterised by pro-inflammatory genes (inc TLR2, Ptgs2, Il12b, Il1b), as well as CD44, Kv1.3, NFkb, Stat1, RelA | [153] |
| Anti-inflammatory disease-associated microglia | Alzheimer’s disease | Mouse | Upregulation of phagocytic genes (inc Igf1, Apoe, Myo1e), as well as CXCR4 and Atf1. | [153] |
| Microglial neurodegenerative phenotype | Alzheimer’s disease | Mouse, human | Loss of 68 homoeostatic genes (inc P2YR12, Tmem119, CX3CR1, CSF1R, TGFBR1) and induction of 28 inflammatory genes (inc CCL2, CSF1, Apoe). TSPO is upregulated on these microglia. | [154] |
| Interferon-related transcriptomic signature microglia | Study analysed a database containing 69 different conditions encompassing neurodegenerative, neoplastic, inflammatory and infectious diseases | Mouse | Dysregulation of many interferon-stimulated genes inc Irf7 and Stat2. Enriched in viral conditions, on LPS-stimulation and in glioma. Also moderately enriched in a number of neurodegenerative disease models. | [152] |
| LPS-related transcriptomic signature microglia | Study analysed a database containing 69 different conditions encompassing neurodegenerative, neoplastic, inflammatory and infectious diseases | Mouse | Upregulation of inflammation-related genes, including Ikbke, cd44, ccl5 and Tspo. Enriched on LPS stimulation and in glioma, and a subset of genes are upregulated in neurodegenerative models (TSPO did not show much change within the LPS signature in the neurodegenerative models). | [152] |
| Neurodegeneration-related transcriptomic signature microglia | Study analysed a database containing 69 different conditions encompassing neurodegenerative, neoplastic, inflammatory and infectious diseases | Mouse | Upregulation of genes that regulate how microglia interact with the environment (inc Bhlhe40, Rxrg, Hif1a and Mitf), and genes that regulate lysosomal function (inc Ctsb, Ctsl and Ctsz). Induced in most neurodegeneration models. | [152] |
| Proliferation-related transcriptomic signature microglia | Study analysed a database containing 69 different conditions encompassing neurodegenerative, neoplastic, inflammatory and infectious diseases | Mouse | Dysregulation of 82 genes associated with proliferation (inc Mki67, Cdk1, Plk1). Enriched in viral or neoplastic-related diseases. | [152] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 3161. https://doi.org/10.3390/ijms20133161
Werry EL, Bright FM, Piguet O, Ittner LM, Halliday GM, Hodges JR, Kiernan MC, Loy CT, Kril JJ, Kassiou M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2019; 20(13):3161. https://doi.org/10.3390/ijms20133161
Chicago/Turabian StyleWerry, Eryn L., Fiona M. Bright, Olivier Piguet, Lars M. Ittner, Glenda M. Halliday, John R. Hodges, Matthew C. Kiernan, Clement T. Loy, Jillian J. Kril, and Michael Kassiou. 2019. "Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders" International Journal of Molecular Sciences 20, no. 13: 3161. https://doi.org/10.3390/ijms20133161
APA StyleWerry, E. L., Bright, F. M., Piguet, O., Ittner, L. M., Halliday, G. M., Hodges, J. R., Kiernan, M. C., Loy, C. T., Kril, J. J., & Kassiou, M. (2019). Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. International Journal of Molecular Sciences, 20(13), 3161. https://doi.org/10.3390/ijms20133161