Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines


Abstract
:1. Introduction
2. Results
2.1. Meta-Analysis on Microarray Expression Data Sets
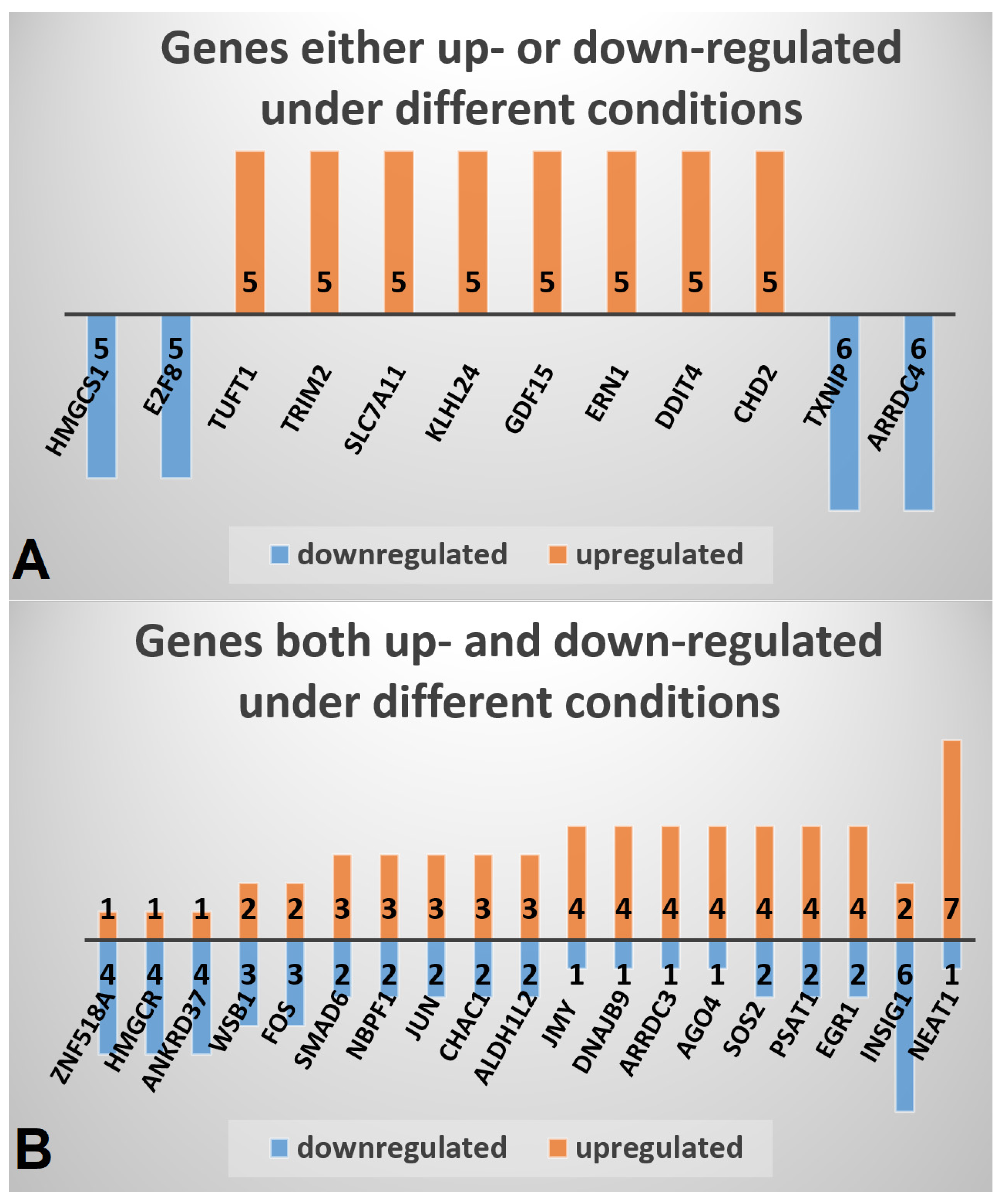
2.2. Genes Either Up- or Down-Regulated in MTF-Treated vs. MTF-Untreated Conditions
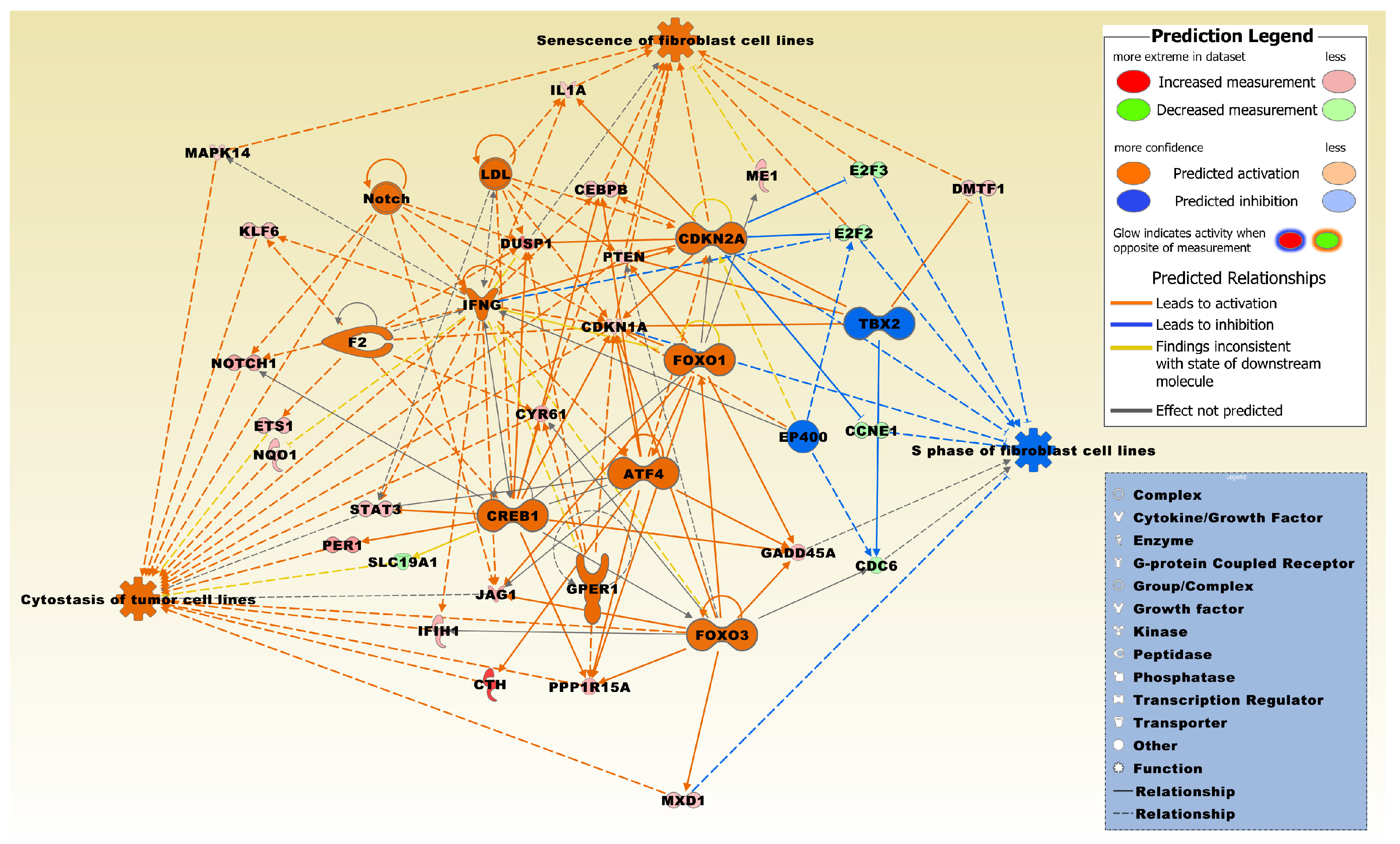
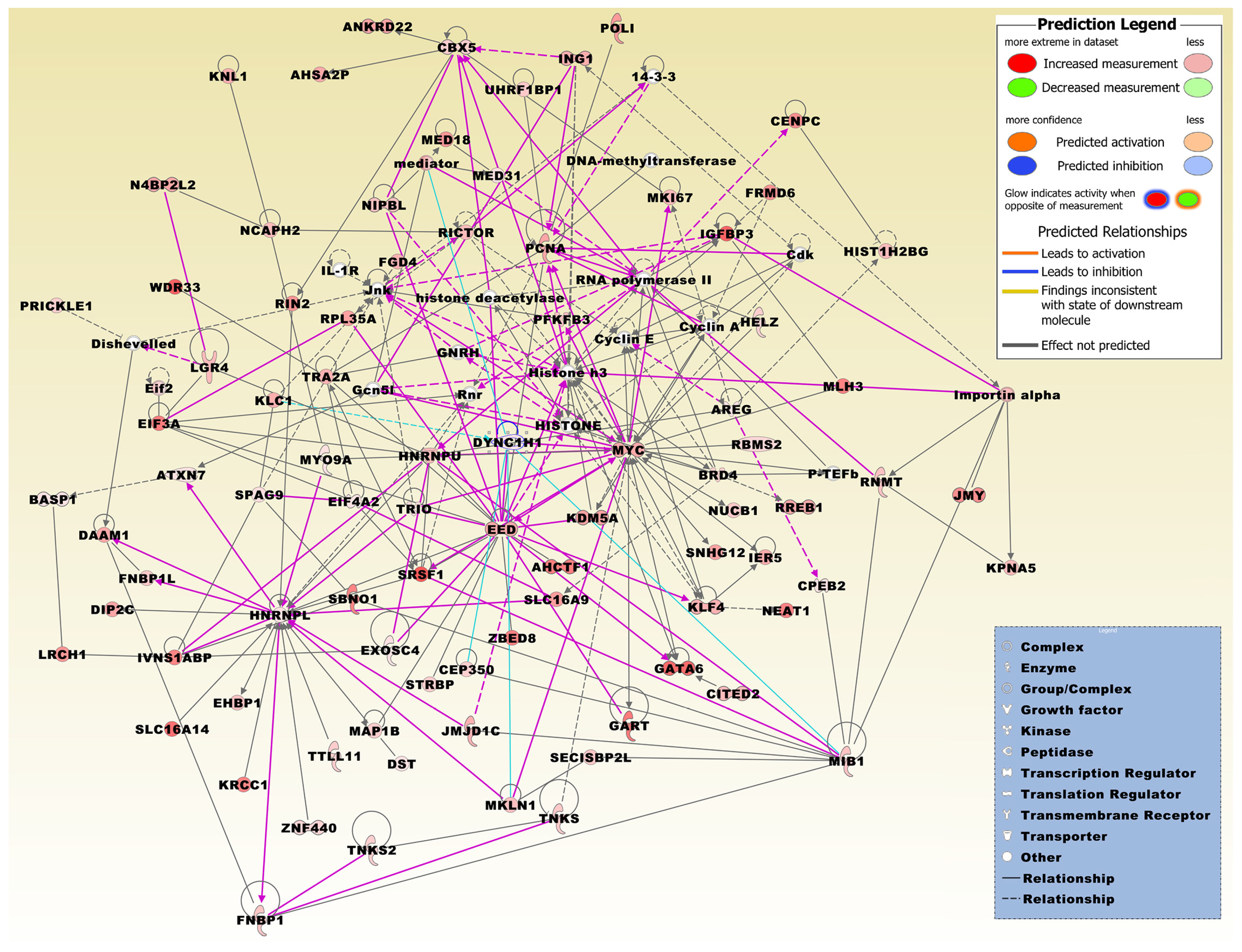
2.3. Biofunctional Analysis on Genes Either Up- or Down-Regulated in MTF-Treated vs. MTF-Untreated Conditions
2.4. Genes Both Up- and Down-Regulated in MTF-Treated vs. MTF-Untreated Conditions
2.5. Biofunctional Analysis of Genes Both Up- and Down-Regulated in MTF-Treated vs. MTF-Untreated Conditions
2.6. DEGs in 6 h and 8 h vs. 24 h MTF Treatment
3. Discussion
3.1. Genes Upregulated under Different Conditions
3.2. Genes Downregulated under Different Conditions
3.3. Genes Both Up- and Down-Regulated under Different Conditions
3.4. DEGs in 6 h and 8 h vs. 24 h MTF Treatment
3.5. Biofunctional Assessment
3.6. Implications for MTF Treatment
4. Materials and Methods
4.1. Selection of Microarray Data Sets
4.2. Calculation of DEGs
4.3. Functional Gene Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| DEG | differentially expressed gene |
| EMT | epithelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| FC | fold change |
| FE | fold enrichment |
| GEO | Gene Expression Omnibus |
| GO | gene ontology |
| MTF | metformin |
| OXPHOS | oxidative phosphorylation |
| T2D | type 2 diabetes |
| UPR | unfolded protein response |
References
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, C.; Gutierrez-Salmeron, M.; Chocarro-Calvo, A.; Garcia-Martinez, J.M.; Castano, A.; De la Vieja, A. From obesity to diabetes and cancer: Epidemiological links and role of therapies. Br. J. Cancer 2016, 114, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Drzewoski, J. Molecular action of metformin in hepatocytes: An updated insight. Curr. Diabetes Rev. 2015, 11, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Czyzyk, A.; Szczepanik, Z. Diabetes mellitus and cancer. Eur. J. Intern. Med. 2000, 11, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, R.; Templeton, A.J.; Seruga, B.; Ocana, A.; Amir, E.; Ethier, J.L. Hyperglycaemia and Survival in Solid Tumours: A Systematic Review and Meta-analysis. Clin. Oncol. (R Coll Radiol) 2018, 30, 215–224. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Wa Cheng, K.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Larsson, O.; Morita, M.; Topisirovic, I.; Alain, T.; Blouin, M.J.; Pollak, M.; Sonenberg, N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA 2012, 109, 8977–8982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Wang, K.; Zheng, N.; Qiu, Y.; Xie, G.; Su, M.; Jia, W.; Li, H. Metformin suppressed the proliferation of LoVo cells and induced a time-dependent metabolic and transcriptional alteration. Sci. Rep. 2015, 5, 17423. [Google Scholar] [CrossRef]
- De Abrew, K.N.; Kainkaryam, R.M.; Shan, Y.K.; Overmann, G.J.; Settivari, R.S.; Wang, X.; Xu, J.; Adams, R.L.; Tiesman, J.P.; Carney, E.W.; et al. Grouping 34 Chemicals Based on Mode of Action Using Connectivity Mapping. Toxicol Sci. 2016, 151, 447–461. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Jennen, D.G.; Magkoufopoulou, C.; Ketelslegers, H.B.; van Herwijnen, M.H.; Kleinjans, J.C.; van Delft, J.H. Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol Sci. 2010, 115, 66–79. [Google Scholar] [CrossRef]
- Schulten, H.J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Semba, Y.; Harada, A.; Maehara, K.; Oki, S.; Meno, C.; Ueda, J.; Yamagata, K.; Suzuki, A.; Onimaru, M.; Nogami, J.; et al. Chd2 regulates chromatin for proper gene expression toward differentiation in mouse embryonic stem cells. Nucleic Acids Res. 2017, 45, 8758–8772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, H.C.; Pare, G.; Hess, S.; Ford, R.J.; Sjaarda, J.; Raman, K.; McQueen, M.; Lee, S.; Haenel, H.; Steinberg, G.R. Growth Differentiation Factor 15 as a Novel Biomarker for Metformin. Diabetes Care 2017, 40, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Anguiano, G.; Lugo-Trampe, J.J.; Camacho, A.; Said-Fernandez, S.; Mercado-Hernandez, R.; Zomosa-Signoret, V.; Rojas-Martinez, A.; Ortiz-Lopez, R. Comparison of specific expression profile in two in vitro hypoxia models. Exp. Ther. Med. 2018, 15, 4777–4784. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) 2018, 38, 12. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Ye, J.; Zhao, F.; Hu, S.; Wang, S. TRIM2 regulates the development and metastasis of tumorous cells of osteosarcoma. Int. J. Oncol. 2018, 53, 1643–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Wang, X.; Wang, T.; Xing, J. TRIM2 downregulation in clear cell renal cell carcinoma affects cell proliferation, migration, and invasion and predicts poor patients’ survival. Cancer Manag Res. 2018, 10, 5951–5964. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Isogaya, K.; Dan, S.; Yamori, T.; Takano, H.; Yao, R.; Morishita, Y.; Taguchi, L.; Morikawa, M.; Heldin, C.H.; et al. TUFT1 interacts with RABGAP1 and regulates mTORC1 signaling. Cell Discov. 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Patwari, P.; Lee, R.T. An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012, 23, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Chai, T.F.; Hong, S.Y.; He, H.; Zheng, L.; Hagen, T.; Luo, Y.; Yu, F.X. A potential mechanism of metformin-mediated regulation of glucose homeostasis: Inhibition of Thioredoxin-interacting protein (Txnip) gene expression. Cell Signal. 2012, 24, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.H.; Kim, Y.; Lee, B.B.; Han, J.; Kim, H.K.; Shim, Y.M.; Kim, D.H. Metformin induces cell cycle arrest at the G1 phase through E2F8 suppression in lung cancer cells. Oncotarget 2017, 8, 101509–101519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, A.; Bozickovic, O.; Bjune, J.I.; Mellgren, G.; Sagen, J.V. Metformin inhibits hepatocellular glucose, lipid and cholesterol biosynthetic pathways by transcriptionally suppressing steroid receptor coactivator 2 (SRC-2). Sci. Rep. 2015, 5, 16430. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Qin, N.; Ren, H.; Yang, M.; Liu, S.; Wang, Q. Metformin Regulating miR-34a Pathway to Inhibit Egr1 in Rat Mesangial Cells Cultured with High Glucose. Int. J. Endocrinol 2018, 2018, 6462793. [Google Scholar] [CrossRef]
- Baron, V.; Adamson, E.D.; Calogero, A.; Ragona, G.; Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene. Ther. 2006, 13, 115–124. [Google Scholar] [CrossRef]
- Gao, S.; Ge, A.; Xu, S.; You, Z.; Ning, S.; Zhao, Y.; Pang, D. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3beta/beta-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J. Exp. Clin. Cancer Res 2017, 36, 179. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, J.; Cai, J.; He, Z.; Yuan, J.; Zhu, X.; Li, Y.; Li, M.; Guan, H. PSAT1 regulates cyclin D1 degradation and sustains proliferation of non-small cell lung cancer cells. Int. J. Cancer 2015, 136, E39–E50. [Google Scholar] [CrossRef]
- García Navas, R.; Nuevo-Tapioles, C.; Liceras-Boillos, P.; Anta, B.; Lillo, C.; Gómez Rodríguez, C.; Baltanas, F.C.; Santos, E. PO-173 Critical requirement of the Sos1 and Sos2 RasGEFs for maintenance of mitochondrial homeostasis. ESMO Open 2018, 3 (Suppl. 2), A295. [Google Scholar]
- Sheffels, E.; Sealover, N.E.; Wang, C.; Kim, D.H.; Vazirani, I.A.; Lee, E.; E, M.T.; Morrison, D.K.; Luo, J.; Kortum, R.L. Oncogenic RAS isoforms show a hierarchical requirement for the guanine nucleotide exchange factor SOS2 to mediate cell transformation. Sci. Signal 2018, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Mardones, G.A.; Burgos, P.V.; Brooks, D.A.; Parkinson-Lawrence, E.; Mattera, R.; Bonifacino, J.S. The trans-Golgi network accessory protein p56 promotes long-range movement of GGA/clathrin-containing transport carriers and lysosomal enzyme sorting. Mol. Biol. Cell 2007, 18, 3486–3501. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Duffin, K.L.; Chintharlapalli, S.; Wu, X. GDF15 and Growth Control. Front Physiol. 2018, 9, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavo, N.; Wurm, R.; Neuhold, S.; Adlbrecht, C.; Vila, G.; Strunk, G.; Clodi, M.; Resl, M.; Brath, H.; Prager, R.; et al. GDF-15 Is Associated with Cancer Incidence in Patients with Type 2 Diabetes. Clin. Chem. 2016, 62, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Morita, M.; Nomura, M.; Tanuma, N. Revisiting glucose metabolism in cancer: Lessons from a PKM knock-in model. Mol. Cell Oncol. 2018, 5, e1472054. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes Metab 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Siegel, P.M.; St-Pierre, J. Metabolic Profiles Associated With Metformin Efficacy in Cancer. Front Endocrinol (Lausanne) 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef]
- Peng, M.; Darko, K.O.; Tao, T.; Huang, Y.; Su, Q.; He, C.; Yin, T.; Liu, Z.; Yang, X. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat Rev. 2017, 54, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Salis, O.; Bedir, A.; Ozdemir, T.; Okuyucu, A.; Alacam, H. The relationship between anticancer effect of metformin and the transcriptional regulation of certain genes (CHOP, CAV-1, HO-1, SGK-1 and Par-4) on MCF-7 cell line. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1602–1609. [Google Scholar] [PubMed]
- Asiedu, M.K.; Barron, M.; Aubry, M.C.; Wigle, D.A. Patient- and Cell Type-Specific Heterogeneity of Metformin Response. Basic Clin. Pharmacol. Toxicol. 2018, 122, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Rustici, G.; Kolesnikov, N.; Brandizi, M.; Burdett, T.; Dylag, M.; Emam, I.; Farne, A.; Hastings, E.; Ison, J.; Keays, M.; et al. ArrayExpress update--trends in database growth and links to data analysis tools. Nucleic Acids Res. 2013, 41, D987–D990. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34(+) cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res 2015, 43, W589–W598. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO 1 Data Set | Cell Lines | Treatment [h] | MTF [mM] | No. of DEGs | Array Platform | Date | Study |
|---|---|---|---|---|---|---|---|
| GSE16816 | SK-4 | 12 | 5 | 101–500 | HumanRef-6 v2.0 expression BeadChips | 2011 | [13] |
| GSE36847 | MCF-7, cytoplasmic | 12 | 10 | 501–1000 | HG-U133 Plus 2.0 | 2012 | [14] |
| GSE36847 | MCF-7, polysome-associated | 12 | 10 | 1001–3000 | HG-U133 Plus 2.0 | 2012 | [14] |
| GSE67342 | LoVo | 8 | 10 | 100–500 | HG-U133 Plus 2.0 | 2015 | [15] |
| GSE67342 | LoVo | 24 | 10 | 1001–3000 | HG-U133 Plus 2.0 | 2015 | [15] |
| GSE69844 | HepaRG | 6 | 0.01 | <100 | HG-U219 | 2016 | [16] |
| GSE69845 | MCF-7 | 6 | 0.01 | <100 | HG-U219 | 2016 | [16] |
| GSE69849 | Ishikawa | 6 | 0.01 | 101–500 | HG-U219 | 2016 | [16] |
| GSE69850 | HepG2 | 6 | 0.01 | 101–500 | HG-U219 | 2016 | [16] |
| GSE97346 | HL60 | 24 | 10 | 1001–3000 | HuGene-2.0 ST | 2017 | [17] |
| GSE97346 | KG1a | 24 | 10 | 101–500 | HuGene-2.0 ST | 2017 | [17] |
| GSE97346 | MOLM14 | 24 | 10 | 101–500 | HuGene-2.0 ST | 2017 | [17] |
| GSE97346 | U937 | 24 | 10 | 501–1000 | HuGene-2.0 ST | 2017 | [17] |
| Category | Genes Either Up- or Down-Regulated | Genes Both Up- and Down-Regulated | ||||
|---|---|---|---|---|---|---|
| p-Value | Overlap [%] | z-Score/Score | p-Value | Overlap [%] | Score | |
| Top canonical pathways | ||||||
| Superpathway of cholesterol biosynthesis | 7.29 × 10−9 | 35.7 | −3.16 | |||
| Estrogen-mediated S phase entry | 1.19 × 10−5 | 26.9 | −2.65 | |||
| Oleate biosynthesis II (animals) | 3.23 × 10−5 | 38.5 | −1.34 | |||
| Cholesterol biosynthesis I | 3.23 × 10−5 | 38.5 | −2.24 | |||
| Mevalonate pathway I | 3.23 × 10−5 | 38.5 | −2.24 | |||
| Prolactin signaling | 5.55 × 10−9 | 14.3 | ||||
| IL-8 signaling | 1.78 × 10−8 | 8.8 | ||||
| HGF signaling | 2.32 × 10−8 | 11.6 | ||||
| PDGF signaling | 1.25 × 10−7 | 12.2 | ||||
| ERBB signaling | 2.99 × 10−7 | 11.3 | ||||
| Top networks related to diseases and functions | ||||||
| Carbohydrate metabolism, small molecule biochemistry, cancer | 58 | |||||
| Cell cycle, cellular assembly and organization, DNA replication, recombination, and repair | 44 | |||||
| Cell cycle, lipid metabolism, molecular transport | 44 | |||||
| RNA post-transcriptional modification, cell cycle, DNA replication, recombination, and repair | 42 | |||||
| Hereditary disorder, neurological disease, organismal injury and abnormalities | 42 | |||||
| RNA post-transcriptional modification, DNA replication, recombination, and repair, cell-to-cell signaling and interaction | 55 | |||||
| Cancer, gastrointestinal disease, organismal injury and abnormalities | 47 | |||||
| Cellular growth and proliferation, post- translational modification, carbohydrate metabolism | 44 | |||||
| RNA post-transcriptional modification, glomerular injury, organismal injury and abnormalities | 40 | |||||
| Cancer, cell cycle, organismal injury and abnormalities | 33 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulten, H.-J.; Bakhashab, S. Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3173. https://doi.org/10.3390/ijms20133173
Schulten H-J, Bakhashab S. Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines. International Journal of Molecular Sciences. 2019; 20(13):3173. https://doi.org/10.3390/ijms20133173
Chicago/Turabian StyleSchulten, Hans-Juergen, and Sherin Bakhashab. 2019. "Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines" International Journal of Molecular Sciences 20, no. 13: 3173. https://doi.org/10.3390/ijms20133173
APA StyleSchulten, H. -J., & Bakhashab, S. (2019). Meta-Analysis of Microarray Expression Studies on Metformin in Cancer Cell Lines. International Journal of Molecular Sciences, 20(13), 3173. https://doi.org/10.3390/ijms20133173