Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of the Salt Inducible Kinase 1 (SIK1) Network in the Response of the Renal Proximal Tubule (RPT) to Injury
2.1. Initial Response of the Renal Proximal Tubule to Injury
2.2. Importance of Na,K-ATPase in Na+ Reabsorption
2.3. SIK Structure and Function
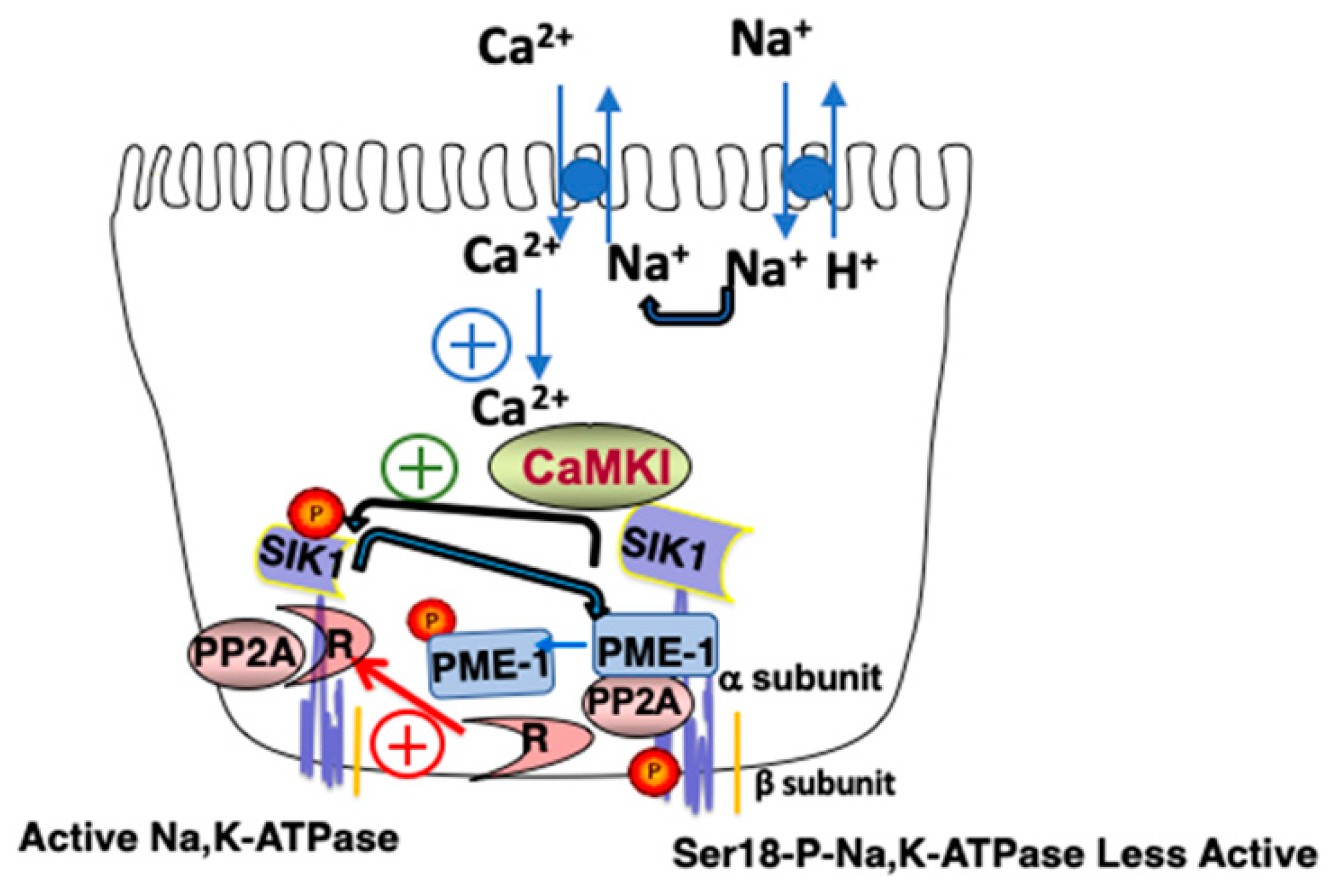
2.4. SIK1 Signaling Network and Na,K-ATPase
2.4.1. Role of SIK1 in Na+ Sensing: Acute Response
2.4.2. Relevance of the SIK1 Signaling Network and Na,K-ATPase to AKI
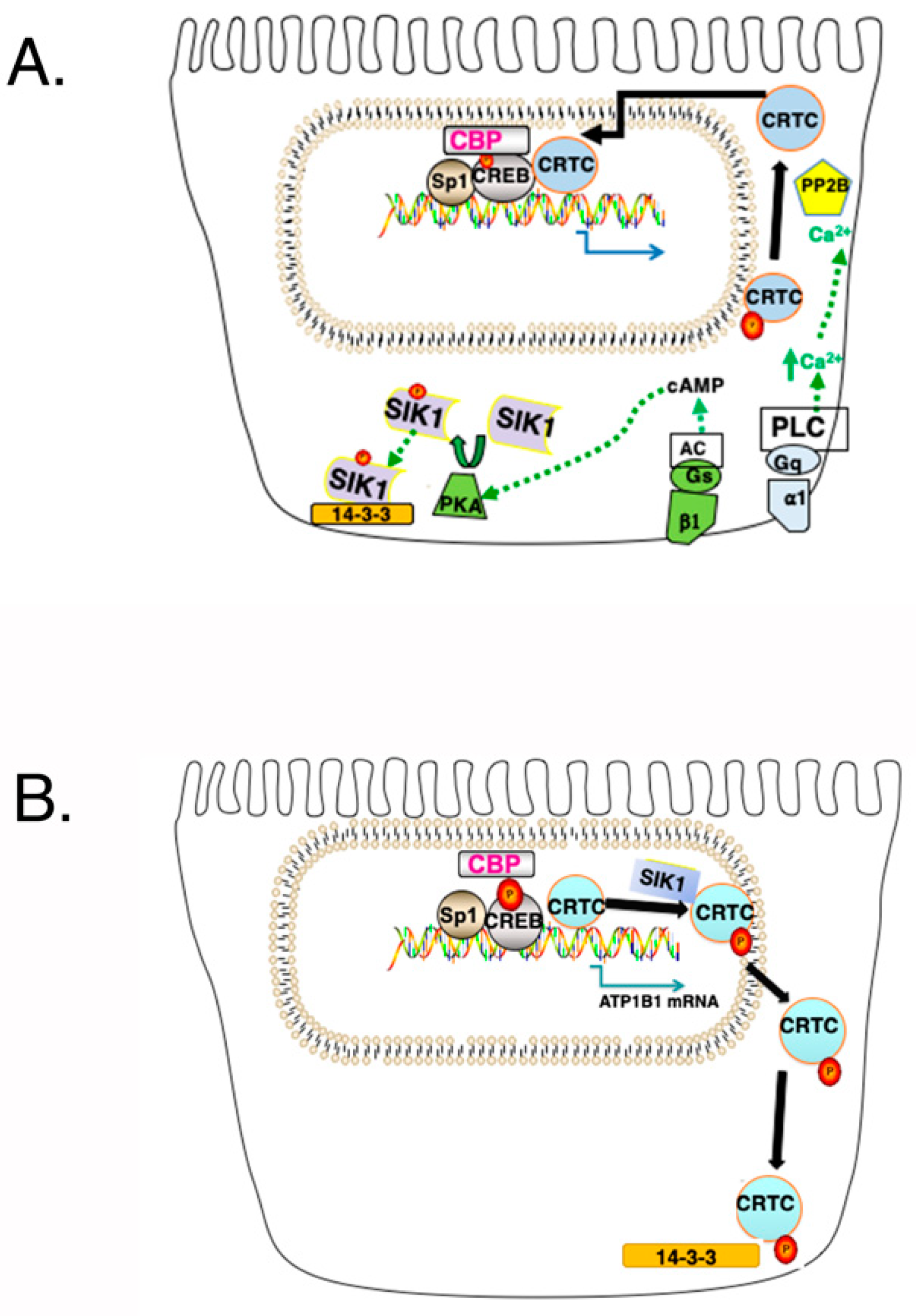
2.5. Transcriptional Effects of SIK1 Involving Its HDAC Kinase Activity and Relevance to AKI
2.6. Involvement of SIK1 in Transcriptional Events Mediated by cAMP and Ca2+
2.7. Summary of the Three Major SIK1 Networks and Their Relevance to Acute Kidney Injury (AKI)
3. Other Roles of SIKs
3.1. Roles of SIK2 and SIK3 in Gluconeogenesis and Lipogenesis
3.1.1. Role of SIK2 and SIK3 in Gluconeogenesis in the Liver
3.1.2. Role of SIK2 and Activating Transcription Factor 3 (ATF3) in Lipid Metabolism in Adipocytes
3.1.3. Relevance to the Kidney
3.2. Role of SIK2 in Mitochondrial Biogenesis and Its Relevance to AKI
3.2.1. Role of SIK2 in Regulating PGC-1α Adipocytes
3.2.2. Regulation of Mitochondrial Biogenesis during AKI
3.3. Yet Other Roles Known to Be Played by SIKs in Events Which Occur during AKI
3.3.1. Fatty Acid Oxidation
ATP Production in the RPT Is Generated by FA Oxidation, Which Declines in AKI
SIK2 Regulates FA Oxidation in Other Tissues
Decreased FA Oxidation Results in EMT in the RPT
Involvement of SIK1 in EMT
3.3.2. Role of SIKs in Apoptosis
3.3.3. Pro-Survival Roles of SIKs and Epigenetics
Prosurvival Role of SIK1 in Myocytes Involves SIK1’s HDAC Kinase Activity
Involvement of Epigenetics and Class IIa HDACs in Regeneration of Damaged Kidneys
3.3.4. Role of SIKs in Cell Growth and Hypertrophy
Cell Division and Hypertrophy during AKI
Role of SIKs in Hypertrophy and Cell Division
4. Summary and Therapeutic Potential
5. Development of Clinical Kinase Drugs
Funding
Acknowledgments
Conflicts of Interest
References
- Okamoto, M.; Takemori, H.; Katoh, Y. Salt-inducible kinase in steroidogenesis and adipogenesis. Trends Endocrinol. Metab. 2004, 15, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Jaitovich, A.; Bertorello, A.M. Intracellular sodium sensing: SIK1 network, hormone action and high blood pressure. Biochim. Et Biophys. Acta 2010, 1802, 1140–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms-Biomarkers-Interventions, and Future Perspectives. Oxid Med. Cell Longev. 2017, 2017, 6193694. [Google Scholar] [CrossRef] [PubMed]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.K.; Hsu, R.K.; Liu, K.D. Management of Acute Kidney Injury: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 72, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Morrell, E.D.; Kellum, J.A.; Hallows, K.R.; Pastor-Soler, N.M. Epithelial transport during septic acute kidney injury. Nephrol. Dial. Transpl. 2014, 29, 1312–1319. [Google Scholar] [CrossRef]
- Strazzullo, P.; Leclercq, C. Sodium. Adv. Nutr. 2014, 5, 188–190. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Cheng, Z.; Qian, Q. Intravenous Fluids and Acute Kidney Injury. Blood Purif. 2017, 43, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, M.; Stenstrom, K.; Eneling, K.; Zwiller, J.; Katz, A.I.; Takemori, H.; Bertorello, A.M. SIK1 is part of a cell sodium-sensing network that regulates active sodium transport through a calcium-dependent process. Proc. Natl. Acad. Sci. USA. 2007, 104, 16922–16927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skou, J.C.; Esmann, M. The Na,K-ATPase. J. Bioenerg. Biomembr. 1992, 24, 249–261. [Google Scholar] [PubMed]
- Sakamoto, K.; Bultot, L.; Goransson, O. The Salt-Inducible Kinases: Emerging Metabolic Regulators. Trends Endocrinol Metab. 2018, 29, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.K.; Satoh, T.; Okamoto, M.; Takemori, H. Importance of autophosphorylation at Ser186 in the A-loop of salt inducible kinase 1 for its sustained kinase activity. J. Cell Biochem. 2008, 104, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Takemori, H.; Lin, X.Z.; Tamura, M.; Muraoka, M.; Satoh, T.; Tsuchiya, Y.; Min, L.; Doi, J.; Miyauchi, A.; et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. Febs. J. 2006, 273, 2730–2748. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, T.; Vaughan, J.M.; Montminy, M. 14-3-3 proteins mediate inhibitory effects of cAMP on salt-inducible kinases (SIKs). Febs. J. 2018, 285, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.; Venetsanou, K.; Chedrese, P.J.; Pinto, V.; Takemori, H.; Franco-Cereceda, A.; Eriksson, P.; Mochizuki, N.; Soares-da-Silva, P.; Bertorello, A.M. Increases in intracellular sodium activate transcription and gene expression via the salt-inducible kinase 1 network in an atrial myocyte cell line, American journal of physiology. Heart Circ. Physiol. 2012, 303, H57–H65. [Google Scholar] [CrossRef]
- Berdeaux, R.; Goebel, N.; Banaszynski, L.; Takemori, H.; Wandless, T.; Shelton, G.D.; Montminy, M. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat. Med. 2007, 13, 597–603. [Google Scholar] [CrossRef]
- Vesely, D.L. Natriuretic peptides and acute renal failure. Am. J. Physiol. Ren. Physiol. 2003, 285, F167–F177. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Lee, J.; Park, J.W.; Hong, J.H.; Kook, H.; Choi, C.; Choi, K.C. Increased expression of atrial natriuretic peptide in the kidney of rats with bilateral ureteral obstruction. Kidney Int. 2001, 59, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, Y.; Sims, T.N.; Urmson, J.; Halloran, P.F. Central role for interferon-gamma receptor in the regulation of renal MHC expression. J. Am. Soc. Nephrol. 2000, 11, 250–261. [Google Scholar] [PubMed]
- Arulkumaran, N.; Prowle, J.R. Natriuretic Peptides: A Role in Early Septic Acute Kidney Injury? Anesthesiology 2018, 129, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Hirahashi, J.; Seki, G.; Tanaka, M.; Kushida, N.; Takeshima, Y.; Nishikawa, Y.; Fujita, T.; Nangaku, M. Successful treatment of acute kidney injury in patients with idiopathic nephrotic syndrome using human atrial natriuretic Peptide. Intern. Med. 2014, 53, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Navaneethan, S.D.; Parikh, C.R.; Hix, J.K. Atrial natriuretic peptide for management of acute kidney injury: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 261–272. [Google Scholar] [CrossRef]
- Wein, M.N.; Foretz, M.; Fisher, D.E.; Xavier, R.J.; Kronenberg, H.M. Salt-Inducible Kinases: Physiology, Regulation by cAMP, and Therapeutic Potential. Trends Endocrinol Metab. 2018, 29, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Garamella, S.; Kim, D.; Rajkhowa, T.; Cutuli, F. Renal Proximal Tubule Na,K-ATPase is Controlled by CREB Regulated Transcriptional CoActivators as well as Salt Inducible Kinase 1. Cell. Signal. 2015, 27, 2568–2578. [Google Scholar] [CrossRef]
- MacKenzie, K.F.; Clark, K.; Naqvi, S.; McGuire, V.A.; Noehren, G.; Kristariyanto, Y.; van den Bosch, M.; Mudaliar, M.; McCarthy, P.C.; Pattison, M.J.; et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 2013, 190, 565–577. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar]
- Breyer, M.D.; Breyer, R.M. Prostaglandin E receptors and the kidney. Am. J. Physiol. Ren. Physiol. 2000, 279, F12–F23. [Google Scholar] [CrossRef]
- Matlhagela, K.; Taub, M. Involvement of EP1 and EP2 receptors in the regulation of the Na,K-ATPase by prostaglandins in MDCK cells. Prostaglandins Other Lipid Mediat. 2006, 79, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, M.B.; Rajkhowa, T.; Cutuli, F.; Springate, J.E.; Taub, M. Regulation of renal proximal tubule Na-K-ATPase by prostaglandins. Am. J. Physiol. Ren. Physiol. 2010, 298, F1222–F1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taub, M.; Borsick, M.; Geisel, J.; Rajkhowa, T.; Allen, C. Regulation of the Na,K-ATPase in MDCK cells by prostaglandin E1: A role for calcium as well as cAMP. Exp. Cell Res. 2004, 299, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. The functional role of the beta-subunit in the maturation and intracellular transport of sodium-potassium ATPase. FEBS Lett. 1991, 285, 189–193. [Google Scholar] [CrossRef]
- Taub, M. Gene Level Regulation of Na,K-ATPase in the Renal Proximal Tubule Is Controlled by Two Independent but Interacting Regulatory Mechanisms Involving Salt Inducible Kinase 1 and CREB-Regulated Transcriptional Coactivators. Int. J. Mol. Sci. 2018, 19, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Borsick, M.; Rajkhowa, T.; Taub, M. Identification of a Prostaglandin-responsive Element in the Na,K-ATPase {beta}1 Promoter That Is Regulated by cAMP and Ca2+: Evidence for an interactive role of cAMP regulatory element-bindin protein and Sp1. J. Biol. Chem. 2005, 280, 334–346. [Google Scholar] [CrossRef]
- Matlhagela, K.; Taub, M. Regulation of the Na-K-ATPase beta(1)-subunit promoter by multiple prostaglandin-responsive elements. Am. J. Physiol. - Ren. Physiol. 2006, 291, F635–F646. [Google Scholar] [CrossRef]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals, Nature reviews. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar]
- Nakajima, T.; Uchida, C.; Anderson, S.F.; Parvin, J.D.; Montminy, M. Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes Dev. 1997, 11, 738–747. [Google Scholar] [CrossRef]
- Bertorello, A.M.; Zhu, J.K. SIK1/SOS2 networks: Decoding sodium signals via calcium-responsive protein kinase pathways. Pflug. Arch. - Eur. J. Physiol. 2009, 458, 613–619. [Google Scholar] [CrossRef]
- Ji, H.; Pardo, J.M.; Batelli, G.; Van Oosten, M.J.; Bressan, R.A.; Li, X. The Salt Overly Sensitive (SOS) pathway: Established and emerging roles. Mol. Plant. 2013, 6, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Sanosaka, M.; Fuchino, H.; Yahara, Y.; Kumagai, A.; Takemoto, D.; Kagawa, M.; Doi, J.; Ohta, M.; Tsumaki, N.; et al. Salt-inducible Kinase 3 Signaling Is Important for the Gluconeogenic Programs in Mouse Hepatocytes. J. Biol. Chem. 2015, 290, 17879–17893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Chen, Q.; Takemori, H.; Xu, H. SIK2 can be activated by deprivation of nutrition and it inhibits expression of lipogenic genes in adipocytes. Obes. (Silver Spring) 2008, 16, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Yates III, J.; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 2008, 456, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yoon, Y.S.; Han, H.S.; Kim, Y.H.; Ogawa, Y.; Park, K.G.; Lee, C.H.; Kim, S.T.; Koo, S.H. SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes 2014, 63, 3659–3673. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sasako, T.; Kubota, N.; Sakurai, Y.; Takamoto, I.; Kubota, T.; Inagi, R.; Seki, G.; Goto, M.; Ueki, K.; et al. Dual Regulation of Gluconeogenesis by Insulin and Glucose in the Proximal Tubules of the Kidney. Diabetes 2017, 66, 2339–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.F.; Cheng, C.F.; Liao, W.J.; Lin, H.; Yang, R.B. ATF3-mediated epigenetic regulation protects against acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Fukushima, A.; Viengchareun, S.; Lombes, M.; Kishi, F.; Miyauchi, A.; Kanematsu, M.; Doi, J.; Kajimura, J.; Nakai, R.; et al. Involvement of SIK2/TORC2 signaling cascade in the regulation of insulin-induced PGC-1alpha and UCP-1 gene expression in brown adipocytes. Am. J. Physiol. Endocrinol Metab. 2009, 296, E1430–E1439. [Google Scholar] [CrossRef] [PubMed]
- Nixon, M.; Stewart-Fitzgibbon, R.; Fu, J.; Akhmedov, D.; Rajendran, K.; Mendoza-Rodriguez, M.G.; Rivera-Molina, Y.A.; Gibson, M.; Berglund, E.D.; Justice, N.J.; et al. Skeletal muscle salt inducible kinase 1 promotes insulin resistance in obesity. Mol. Metab. 2016, 5, 34–46. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA. 2006, 103, 14379–14384. [Google Scholar] [CrossRef]
- Rasbach, K.A.; Schnellmann, R.G. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 2007, 355, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharm. Exp. 2010, 333, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.B.; Gibbs, W.S.; Miller, S.R.; Dupre, T.V.; Megyesi, J.; Beeson, C.C.; Schnellmann, R.G. Proximal Tubule beta 2-Adrenergic Receptor Mediates Formoterol-Induced Recovery of Mitochondrial and Renal Function after Ischemia-Reperfusion Injury. J. Pharm. Exp. 2019, 369, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. (Lausanne) 2015, 2, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 431–436. [Google Scholar] [CrossRef]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Gulati, S.; Orak, J.K.; Singh, A.K. Expression of antioxidant enzymes in rat kidney during ischemia-reperfusion injury. Mol. Cell Biochem. 1993, 125, 97–104. [Google Scholar] [CrossRef]
- Tran, M.; Parikh, S.M. Mitochondrial biogenesis in the acutely injured kidney. Nephron Clin. Pr. 2014, 127, 42–45. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; Dadras, M.S.; Lomnytska, M.; Mahzabin, T.; Lee Miller, M.; Busch, C.; Brunak, S.; Heldin, C.H.; Moustakas, A. The protein kinase SIK downregulates the polarity protein Par3. Oncotarget 2018, 9, 5716–5735. [Google Scholar] [CrossRef]
- Goodwin, J.M.; Svensson, R.U.; Lou, H.J.; Winslow, M.M.; Turk, B.E.; Shaw, R.J. An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol Cell 2014, 55, 436–450. [Google Scholar] [CrossRef]
- Gewin, L.S. Transforming Growth Factor-beta in the Acute Kidney Injury to Chronic Kidney Disease Transition. Nephron 2019, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Havasi, A.; Borkan, S.C. Apoptosis and acute kidney injury. Kidney Int. 2011, 80, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Liu, P.; Wang, Z.C.; Zou, L.; Santiago, S.; Garbitt, V.; Gjoerup, O.V.; Iglehart, J.D.; Miron, A.; Richardson, A.L.; et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci. Signal. 2009, 2, ra35. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Birdsey, N.; Huggins, G.S.; Svensson, E.; Heppe, D.; Knaub, L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2056–H2068. [Google Scholar] [CrossRef] [Green Version]
- Jhala, U.S.; Canettieri, G.; Screaton, R.A.; Kulkarni, R.N.; Krajewski, S.; Reed, J.; Walker, J.; Lin, X.; White, M.; Montminy, M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003, 17, 1575–1580. [Google Scholar] [CrossRef]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2011, 116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Akhmedov, D.; Robb, C.; Leiter, C.; Berdeaux, R. Regulation of SIK1 abundance and stability is critical for myogenesis. Proc. Natl. Acad. Sci. USA. 2013, 110, 117–122. [Google Scholar] [CrossRef]
- Dressler, G.R.; Patel, S.R. Epigenetics in kidney development and renal disease. Transl. Res. 2015, 165, 166–176. [Google Scholar] [CrossRef]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Fujita, T. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J. Am. Soc. Nephrol. 2008, 19, 1311–1320. [Google Scholar] [CrossRef]
- Thomasova, D.; Anders, H.J. Cell cycle control in the kidney. Nephrol. Dial. Transpl. 2015, 30, 1622–1630. [Google Scholar] [CrossRef]
- Wu, C.F.; Chiang, W.C.; Lai, C.F.; Chang, F.C.; Chen, Y.T.; Chou, Y.H.; Wu, T.H.; Linn, G.R.; Ling, H.; Wu, K.D.; et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.; Takemori, H.; Tokudome, T.; Mao, Y.; Otani, K.; Mochizuki, N.; Pires, N.; Pinho, M.J.; Franco-Cereceda, A.; Torielli, L.; et al. Lack of salt-inducible kinase 2 (SIK2) prevents the development of cardiac hypertrophy in response to chronic high-salt intake. PLoS ONE 2014, 9, e95771. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, S.; Takemori, H.; Uebi, T.; Ikegami, D.; Hiramatsu, K.; Ikegawa, S.; Yoshikawa, H.; Tsumaki, N. SIK3 is essential for chondrocyte hypertrophy during skeletal development in mice. Development 2012, 139, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, F.; Mannion, D.; Liu, S.; Zheng, Y.; Mangala, L.S.; Redondo, C.; Herrero-Gonzalez, S.; Xu, R.; Taylor, C.; Chedom, D.F.; et al. Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer Cell 2016, 30, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Lu, Z.; Jennings, N.B.; Etemadmoghadam, D.; Capalbo, L.; Jacamo, R.O.; Barbosa-Morais, N.; Le, X.F.; Australian Ovarian Cancer Study Group; Vivas-Mejia, P.; et al. SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer. Cancer Cell 2010, 18, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Wehr, M.C.; Holder, M.V.; Gailite, I.; Saunders, R.E.; Maile, T.M.; Ciirdaeva, E.; Instrell, R.; Jiang, M.; Howell, M.; Rossner, M.J.; et al. Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat. Cell Biol. 2013, 15, 61–71. [Google Scholar] [CrossRef]
- Chen, H.; Huang, S.; Han, X.; Zhang, J.; Shan, C.; Tsang, Y.H.; Ma, H.T.; Poon, R.Y. Salt-inducible kinase 3 is a novel mitotic regulator and a target for enhancing antimitotic therapeutic-mediated cell death. Cell Death Dis. 2014, 5, e1177. [Google Scholar] [CrossRef]
- Li, Y.; Xia, W.; Zhao, F.; Wen, Z.; Zhang, A.; Huang, S.; Jia, Z.; Zhang, Y. Prostaglandins in the pathogenesis of kidney diseases. Oncotarget 2018, 9, 26586–26602. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Sugiura, H.; Mitobe, M.; Tsuchiya, K.; Shirota, S.; Nishimura, S.; Shiohira, S.; Ito, H.; Nobori, K.; Gullans, S.R.; et al. ATF3 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 217–224. [Google Scholar] [CrossRef]
- Chen, F.; Chen, L.; Qin, Q.; Sun, X. Salt-Inducible Kinase 2: An Oncogenic Signal Transmitter and Potential Target for Cancer Therapy. Front. Oncol. 2019, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Foretz, M.; Marion, A.; Campbell, D.G.; Gourlay, R.; Boudaba, N.; Tournier, E.; Titchenell, P.; Peggie, M.; Deak, M.; et al. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat. Commun. 2014, 5, 4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, T.; Fujiyama, T.; Miyoshi, C.; Ikkyu, A.; Hotta-Hirashima, N.; Kanno, S.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; Funato, H.; et al. A single phosphorylation site of SIK3 regulates daily sleep amounts and sleep need in mice. Proc. Natl. Acad. Sci. USA. 2018, 115, 10458–10463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, P. Kidney-specific gene targeting. J. Am. Soc. Nephrol. 2004, 15, 2237–2239. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, T.B.; Choi, H.G.; Song, J.H.; Russell, C.N.; Hussain, M.M.; Graham, D.B.; Khor, B.; Gagnon, J.; O’Connell, D.J.; Narayan, K.; et al. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proc. Natl. Acad. Sci. USA. 2014, 111, 12468–12473. [Google Scholar] [CrossRef] [Green Version]
- Heap, R.E.; Hope, A.G.; Pearson, L.A.; Reyskens, K.; McElroy, S.P.; Hastie, C.J.; Porter, D.W.; Arthur, J.S.C.; Gray, D.W.; Trost, M. Identifying Inhibitors of Inflammation: A Novel High-Throughput MALDI-TOF Screening Assay for Salt-Inducible Kinases (SIKs). Slas. Discov. 2017, 22, 1193–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taub, M. Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 3219. https://doi.org/10.3390/ijms20133219
Taub M. Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential. International Journal of Molecular Sciences. 2019; 20(13):3219. https://doi.org/10.3390/ijms20133219
Chicago/Turabian StyleTaub, Mary. 2019. "Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential" International Journal of Molecular Sciences 20, no. 13: 3219. https://doi.org/10.3390/ijms20133219
APA StyleTaub, M. (2019). Salt Inducible Kinase Signaling Networks: Implications for Acute Kidney Injury and Therapeutic Potential. International Journal of Molecular Sciences, 20(13), 3219. https://doi.org/10.3390/ijms20133219