Multi-Faceted Notch in Allergic Airway Inflammation

Abstract
:1. Introduction
1.1. Allergic Airway Diseases
1.2. Notch Signaling Pathway
1.3. Notch in Lung Development
2. Notch Signaling in Acute Allergic Airway Inflammation
2.1. Notch in Th Subset Differentiation and Immune Responses
2.2. Notch Ligand-Specific Allergic Immune Responses
2.3. Contradictory Findings
3. The Role of Notch Signaling in Chronic Allergic Airway Inflammation
3.1. Airway Remodeling and Angiogenesis in Chronic Asthma
3.2. Notch Signaling Family and Angiogenesis
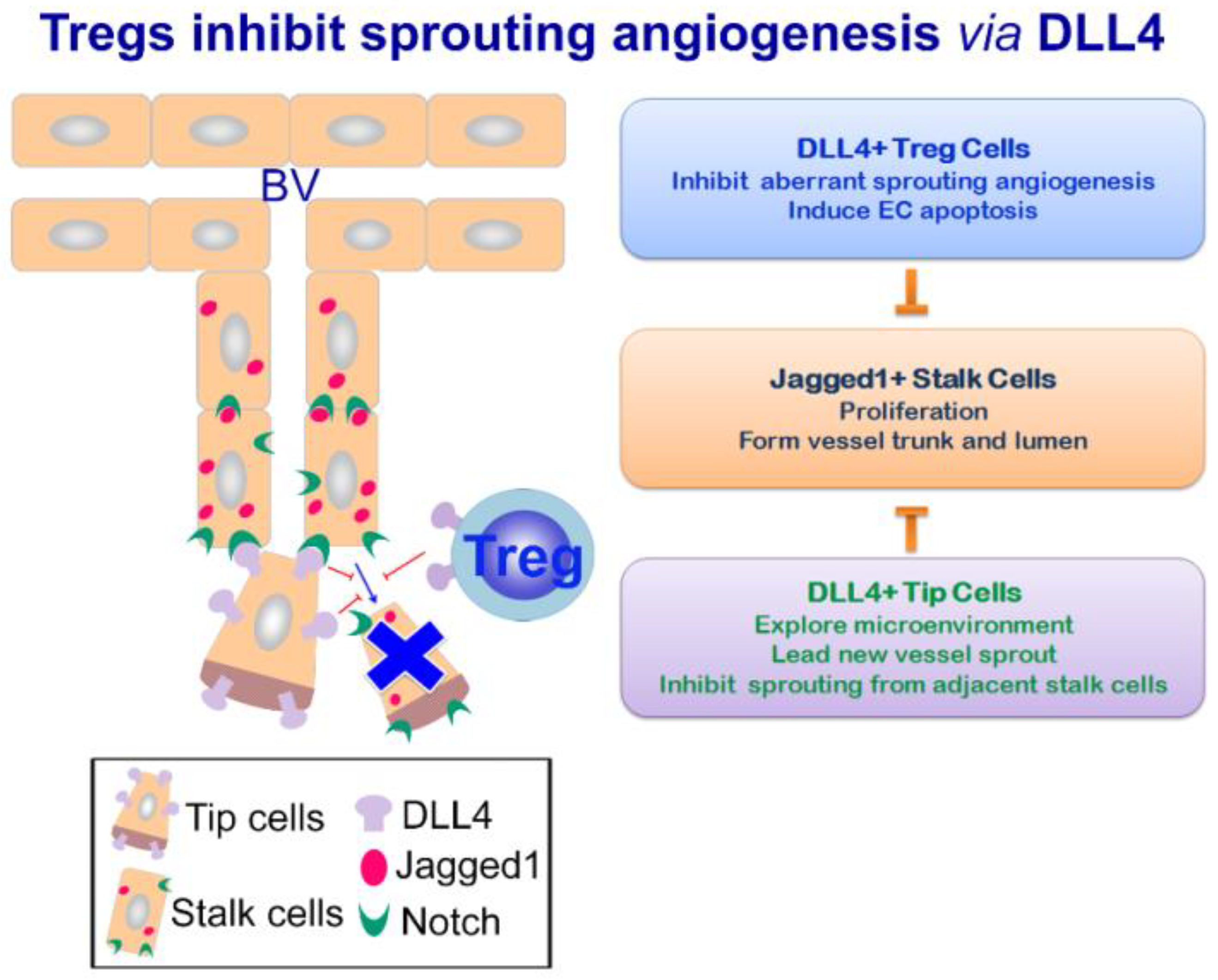
3.3. Tregs Ameliorate Remodeling Angiogenesis by DLL4-Notch Signaling
4. Conclusions
Funding
Conflicts of Interest
References
- Vahedi, G.; Richard, A.C.; O’Shea, J.J. Enhancing the understanding of asthma. Nat. Immunol. 2014, 15, 701–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umetsu, D.T.; McIntire, J.J.; Akbari, O.; Macaubas, C.; DeKruyff, R.H. Asthma: An epidemic of dysregulated immunity. Nat. Immunol. 2002, 3, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.H.; Judd, S.; Thomas, J.A.; Larcombe, A.N.; Sly, P.D.; Holt, P.G. Boosting airway T-regulatory cells by gastrointestinal stimulation as a strategy for asthma control. Mucosal Immunol. 2011, 4, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Barker, J.E.; Robinson, D.S.; Lloyd, C.M. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 2005, 202, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Lewkowich, I.P.; Herman, N.S.; Schleifer, K.W.; Dance, M.P.; Chen, B.L.; Dienger, K.M.; Sproles, A.A.; Shah, J.S.; Kohl, J.; Belkaid, Y.; et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J. Exp. Med. 2005, 202, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.H.; Stumbles, P.A.; Zosky, G.R.; Subrata, L.S.; Thomas, J.A.; Turner, D.J.; Sly, P.D.; Holt, P.G. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J. Exp. Med. 2006, 203, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma-present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Barnes, P.J. Pathophysiology of allergic inflammation. Immunol. Rev. 2011, 242, 31–50. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Leavy, O. Asthma and allergy: An IFN-gamma bias in severe asthma. Nat. Rev. Immunol. 2015, 15, 466–467. [Google Scholar] [CrossRef]
- Shamji, M.H.; Durham, S.R. Mechanisms of allergen immunotherapy for inhaled allergens and predictive biomarkers. J. Allergy Clin. Immunol. 2017, 140, 1485–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzukawa, M.; Ohta, K.; Nagase, H.; Ohta, S. Antibody therapy for the management of severe asthma with eosinophilic inflammation. Int. Immunol. 2017, 29, 337–343. [Google Scholar] [CrossRef]
- Papathanassiou, E.; Loukides, S.; Bakakos, P. Severe asthma: Anti-IgE or anti-IL-5? Eur. Clin. Respir. J. 2016, 3, 31813. [Google Scholar] [CrossRef] [PubMed]
- Van de Veen, W.; Akdis, M. The use of biologics for immune modulation in allergic disease. J. Clin. Investig. 2019, 129, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajt, M.L.; Wenzel, S.E. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: The next steps toward personalized care. J. Allergy Clin. Immunol. 2015, 135, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Matucci, A.; Vultaggio, A.; Maggi, E.; Kasujee, I. Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respir. Res. 2018, 19, 113. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Srivastava, D. Heart disease modelling adds a Notch to its belt. Nat. Cell Biol. 2016, 18, 3–5. [Google Scholar] [CrossRef]
- D’Amato, G.; Luxan, G.; del Monte-Nieto, G.; Martinez-Poveda, B.; Torroja, C.; Walter, W.; Bochter, M.S.; Benedito, R.; Cole, S.; Martinez, F.; et al. Sequential Notch activation regulates ventricular chamber development. Nat. Cell Biol. 2016, 18, 7–20. [Google Scholar] [CrossRef]
- Vieira, N.M.; Elvers, I.; Alexander, M.S.; Moreira, Y.B.; Eran, A.; Gomes, J.P.; Marshall, J.L.; Karlsson, E.K.; Verjovski-Almeida, S.; Lindblad-Toh, K.; et al. Jagged 1 rescues the Duchenne muscular dystrophy phenotype. Cell 2015, 163, 1204–1213. [Google Scholar] [CrossRef]
- Obata, Y.; Takahashi, D.; Ebisawa, M.; Kakiguchi, K.; Yonemura, S.; Jinnohara, T.; Kanaya, T.; Fujimura, Y.; Ohmae, M.; Hase, K.; et al. Epithelial cell-intrinsic Notch signaling plays an essential role in the maintenance of gut immune homeostasis. J. Immunol. 2012, 188, 2427–2436. [Google Scholar] [CrossRef]
- Tsao, P.-N.; Matsuoka, C.; Wei, S.-C.; Sato, A.; Sato, S.; Hasegawa, K.; Chen, H.-K.; Ling, T.-Y.; Mori, M.; Cardoso, W.V.; et al. Epithelial Notch signaling regulates lung alveolar morphogenesis and airway epithelial integrity. Proc. Natl. Acad. Sci. USA 2016, 113, 8242–8247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, D.; Ouyang, R.; Li, J.; Chen, Y.; Chen, P. Notch signaling in lung diseases: Focus on Notch1 and Notch3. Ther. Adv. Respir. Dis. 2016, 10, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Xu, C.; Ahmad, M.; Yang, Y.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Notch signaling: Linking embryonic lung development and asthmatic airway remodeling. Mol. Pharmacol. 2017, 92, 676–693. [Google Scholar] [CrossRef] [PubMed]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol. Metab. TEM 2015, 26, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; mbesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Moghal, N.; Egan, S.E. Notch signaling in lung development and disease. In Notch Signaling in Embryology and Cancer; Reichrath, J., Reichrath, S., Eds.; Springer: New York, NY, USA, 2012; pp. 89–98. [Google Scholar]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722. [Google Scholar] [CrossRef]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Kidd, S.; Kelley, M.R.; Young, M.W. Sequence of the notch locus of Drosophila melanogaster: Relationship of the encoded protein to mammalian clotting and growth factors. Mol. Cell. Biol. 1986, 6, 3094–3108. [Google Scholar] [CrossRef]
- Struhl, G.; Fitzgerald, K.; Greenwald, I. Intrinsic activity of the lin-12 and Notch intracellular domains in vivo. Cell 1993, 74, 331–345. [Google Scholar] [CrossRef]
- Struhl, G.; Adachi, A. Nuclear access and action of Notch in vivo. Cell 1998, 93, 649–660. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, I. Notch and the awesome power of genetics. Genetics 2012, 191, 655–669. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Chapter three—Canonical and non-canonical Notch ligands. In Current Topics in Developmental Biology; Kopan, R., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 92, pp. 73–129. [Google Scholar]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Helbig, C.; Backer, R.A. Notch in T cell differentiation: All things considered. Trends Immunol. 2015, 36, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Blander, J.M.; Lee, G.R.; Tanigaki, K.; Honjo, T.; Flavell, R.A. Instruction of distinct CD4 T helper cell fates by different Notch ligands on antigen-presenting cells. Cell 2004, 117, 515–526. [Google Scholar] [CrossRef]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic ligand discrimination in the Notch signaling pathway. Cell 2018, 172, 869–880. [Google Scholar] [CrossRef]
- Hicks, C.; Johnston, S.H.; diSibio, G.; Collazo, A.; Vogt, T.F.; Weinmaster, G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2000, 2, 515–520. [Google Scholar] [CrossRef]
- Yang, L.T.; Nichols, J.T.; Yao, C.; Manilay, J.O.; Robey, E.A.; Weinmaster, G. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol. Biol. Cell 2005, 16, 927–942. [Google Scholar] [CrossRef]
- Morimoto, M.; Nishinakamura, R.; Saga, Y.; Kopan, R. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development 2012, 139, 4365–4373. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Connett, J.M.; Kunkel, S.L.; Matsukawa, A. Notch system in the linkage of innate and adaptive immunity. J. Leukoc. Biol. 2012, 92, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Janke, M.; Kassner, N.; Hohnstein, T.; Krueger, M.; Scheffold, A. Notch regulates IL-10 production by T helper 1 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3497–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassner, N.; Krueger, M.; Yagita, H.; Dzionek, A.; Hutloff, A.; Kroczek, R.; Scheffold, A.; Rutz, S. Cutting edge: Plasmacytoid dendritic cells induce IL-10 production in T cells via the delta-like-4/Notch axis. J. Immunol. 2010, 184, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.-A.; Schaller, M.A.; de Almeida Nagata, D.E.; Rasky, A.J.; Maillard, I.P.; Lukacs, N.W. Notch ligand delta-like 4 promotes regulatory T cell identity in plmonary viral infection. J. Immunol. 2017, 198, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Schaller, M.; Hogaboam, C.M.; Standiford, T.J.; Sandor, M.; Lukacs, N.W.; Chensue, S.W.; Kunkel, S.L. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J. Clin. Investig. 2009, 119, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Hossain, F.; Pannuti, A.; Lessard, C.B.; Ladd, G.Z.; Jung, J.I.; Minter, L.M.; Osborne, B.A.; Miele, L.; Golde, T.E. γ-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol. Med. 2017, 9, 950–966. [Google Scholar] [CrossRef]
- Gu, K.; Li, Q.; Lin, H.; Zhu, J.; Mo, J.; He, S.; Lu, X.; Jiang, X.; Sun, H. Gamma secretase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2017, 27, 851–866. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, B.S.; Uhm, T.G.; Lee, S.H.; Lee, G.R.; Park, C.S.; Chung, I.Y. Gamma-secretase inhibitor reduces allergic pulmonary inflammation by modulating Th1 and Th2 responses. Am. J. Respir. Crit. Care Med. 2009, 179, 875–882. [Google Scholar] [CrossRef]
- Okamoto, M.; Matsuda, H.; Joetham, A.; Lucas, J.J.; Domenico, J.; Yasutomo, K.; Takeda, K.; Gelfand, E.W. Jagged1 on dendritic cells and Notch on CD4+ T cells initiate lung allergic responsiveness by inducing IL-4 production. J. Immunol. 2009, 183, 2995–3003. [Google Scholar] [CrossRef]
- Okamoto, M.; Takeda, K.; Joetham, A.; Ohnishi, H.; Matsuda, H.; Swasey, C.H.; Swanson, B.J.; Yasutomo, K.; Dakhama, A.; Gelfand, E.W. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J. Exp. Med. 2008, 205, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Vroman, H.; Lukkes, M.; van Nimwegen, M.; de Bruijn, M.J.W.; Li, B.W.S.; Kleinjan, A.; de Boer, G.M.; Tramper-Stranders, G.A.; Kool, M.; et al. Increased surface expression of NOTCH on memory T cells in peripheral blood from patients with asthma. J. Allergy Clin. Immunol. 2019, 143, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Radke, A.L.; Reynolds, L.E.; Melo, R.C.N.; Dvorak, A.M.; Weller, P.F.; Spencer, L.A. Mature human eosinophils express functional Notch ligands mediating eosinophil autocrine regulation. Blood 2009, 113, 3092–3101. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Wang, H.; Xenakis, J.J.; Spencer, L.A. Notch signaling mediates granulocyte-macrophage colony-stimulating factor priming-induced transendothelial migration of human eosinophils. Allergy 2015, 70, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.F.; Abdala-Valencia, H.; Anekalla, K.R.; Cuervo-Pardo, L.; Gottardi, C.J.; Berdnikovs, S. Beyond epithelial-to-mesenchymal transition: Common suppression of differentiation programs underlies epithelial barrier dysfunction in mild, moderate, and severe asthma. Allergy 2017, 72, 1988–2004. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.A.E.; Vonk, J.M.; Himes, B.E.; Sarnowski, C.; Minelli, C.; Jarvis, D.; Bouzigon, E.; Nickle, D.C.; Laviolette, M.; Sin, D.; et al. PTTG1IP and MAML3, novel genomewide association study genes for severity of hyperresponsiveness in adult asthma. Allergy 2017, 72, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Antov, A.; Flavell, R.A. The different faces of Notch in T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Phipps, S.; Angkasekwinai, P.; Dong, C.; Foster, P.S. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 2010, 185, 4681–4690. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.M.; Chen, H.C.; Pochard, P.; Eisenbarth, S.C.; Herrick, C.A.; Bottomly, H.K. TLR4 signaling in stromal cells is critical for the initiation of allergic Th2 responses to inhaled antigen. J. Immunol. 2010, 184, 3535–3544. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Oriss, T.B.; Paglia, M.; Fei, M.; Yarlagadda, M.; Vanhaesebroeck, B.; Ray, A.; Ray, P. Activation of c-Kit in dendritic cells regulates T helper cell differentiation and allergic asthma. Nat. Med. 2008, 14, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Gomi, K.; Staudt, M.R.; Salit, J.; Kaner, R.J.; Heldrich, J.; Rogalski, A.M.; Arbelaez, V.; Crystal, R.G.; Walters, M.S. JAG1-mediated Notch signaling regulates secretory cell differentiation of the human airway epithelium. Stem Cell Rev. Rep. 2016, 12, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Krawczyk, C.J.; Pearce, E.J. Suppression of Th2 cell development by Notch ligands Delta1 and Delta4. J. Immunol. 2008, 180, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Bradshaw, E.M.; Wang, Y.; Oukka, M.; Kivisakk, P.; Chiba, S.; Yagita, H.; Khoury, S.J. Jagged1 and Delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 5990–5998. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Sumi, T.; Ishida, W.; Ojima, A.; Kajisako, M.; Koyanagi, A.; Koyama, N.; Yagita, H. Notch ligand Delta-like4 inhibits the development of murine experimental allergic conjunctivitis. Immunol. Lett. 2008, 121, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Schaller, M.; Berlin, A.A.; Lukacs, N.W. Notch ligand Delta-like 4 regulates development and pathogenesis of allergic airway responses by modulating IL-2 production and Th2 immunity. J. Immunol. 2010, 185, 5835–5844. [Google Scholar] [CrossRef]
- Schaller, M.A.; Neupane, R.; Rudd, B.D.; Kunkel, S.L.; Kallal, L.E.; Lincoln, P.; Lowe, J.B.; Man, Y.; Lukacs, N.W. Notch ligand Delta-like 4 regulates disease pathogenesis during respiratory viral infections by modulating Th2 cytokines. J. Exp. Med. 2007, 204, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.-T.; Chen, Y.-L.; Lien, C.-I.; Liu, W.-L.; Hsu, L.-C.; Yagita, H.; Chiang, B.-L. Notch ligand DLL4 alleviates allergic airway inflammation via induction of a homeostatic regulatory pathway. Sci. Rep. 2017, 7, 43535. [Google Scholar] [CrossRef]
- Hoyne, G.F.; Dallman, M.J.; Lamb, J.R. Linked suppression in peripheral T cell tolerance to the house dust mite derived allergen Der p 1. Int. Arch. Allergy Immunol. 1999, 118, 122–124. [Google Scholar] [CrossRef]
- Mukherjee, S.; Schaller, M.A.; Neupane, R.; Kunkel, S.L.; Lukacs, N.W. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J. Immunol. 2009, 182, 7381–7388. [Google Scholar] [CrossRef]
- Shibata, K.; Yamada, H.; Sato, T.; Dejima, T.; Nakamura, M.; Ikawa, T.; Hara, H.; Yamasaki, S.; Kageyama, R.; Iwakura, Y.; et al. Notch-Hes1 pathway is required for the development of IL-17-producing gd T cells. Blood 2011, 118, 586–593. [Google Scholar] [CrossRef]
- Zaman, T.S.; Arimochi, H.; Maruyama, S.; Ishifune, C.; Tsukumo, S.-i.; Kitamura, A.; Yasutomo, K. Notch balances Th17 and induced regulatory T cell functions in dendritic cells by regulating Aldh1a2 expression. J. Immunol. 2017, 199, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Wakashin, H.; Hirose, K.; Maezawa, Y.; Kagami, S.I.; Suto, A.; Watanabe, N.; Saito, Y.; Hatano, M.; Tokuhisa, T.; Iwakura, Y.; et al. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2008, 178, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Melton, A.C.; Chen, C.; Engler, M.B.; Huang, K.E.; Ren, X.; Wang, Y.; Bernstein, X.; Li, J.T.; Atabai, K.; et al. IL-17A produced by [alpha][beta] T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 2012, 18, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Irving, K. Asthma and allergy: Complementing the IL-17 axis in asthma. Nat. Rev. Immunol. 2010, 10, 676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Sheng, A.; Weng, C.; Zhu, T.; Zhao, W.; Li, C. Gamma-secretase inhibitor alleviates acute airway inflammation of allergic asthma in mice by downregulating Th17 cell differentiation. Mediat. Inflamm. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Weng, C.; Chong, L.; Jia, X.; Zheng, R.; Huang, Y.; Zhu, T.; Li, C.; Zhang, W. Anti-Dll4 antibody inhibits the differentiation of Th17 cells in asthmatic mice. Inflammation 2017, 40, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sheng, A.; Jia, X.; Zeng, Z.; Zhang, X.; Zhao, W.; Zhang, W. Th17/Treg dysregulation in allergic asthmatic children is associated with elevated notch expression. J. Asthma 2018, 55, 1–7. [Google Scholar] [CrossRef]
- Nakano, N.; Nishiyama, C.; Yagita, H.; Koyanagi, A.; Akiba, H.; Chiba, S.; Ogawa, H.; Okumura, K. Notch signaling confers antigen-presenting cell functions on mast cells. J. Allergy Clin. Immunol. 2009, 123, 74–81. [Google Scholar] [CrossRef]
- Murata, A.; Okuyama, K.; Sakano, S.; Kajiki, M.; Hirata, T.; Yagita, H.; Zuniga-Pflucker, J.C.; Miyake, K.; kashi-Takamura, S.; Moriwaki, S.; et al. A Notch ligand, Delta-like 1 functions as an adhesion molecule for mast cells. J. Immunol. 2010, 185, 3905–3912. [Google Scholar] [CrossRef]
- Bassil, R.; Zhu, B.; Lahoud, Y.; Riella, L.V.; Yagita, H.; Elyaman, W.; Khoury, S.J. Notch ligand Delta-like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J. Immunol. 2011, 187, 2322–2328. [Google Scholar] [CrossRef]
- Billiard, F.; Lobry, C.; Darrasse-Jeze, G.; Waite, J.; Liu, X.; Mouquet, H.; DaNave, A.; Tait, M.; Idoyaga, J.; Leboeuf, M.; et al. Dll4-Notch signaling in Flt3-independent dendritic cell development and autoimmunity in mice. J. Exp. Med. 2012, 209, 1011–1028. [Google Scholar] [CrossRef] [PubMed]
- Tran, I.T.; Sandy, A.R.; Carulli, A.J.; Ebens, C.; Chung, J.; Shan, G.T.; Radojcic, V.; Friedman, A.; Gridley, T.; Shelton, A.; et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J. Clin. Investig. 2013, 123, 1590–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordon, Y. T cell responses: Jagged gives an edge to TH1 cells. Nat. Rev. Immunol. 2012, 12, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Le Friec, G.; Sheppard, D.; Whiteman, P.; Karsten, C.M.; Shamoun, S.A.-T.; Laing, A.; Bugeon, L.; Dallman, M.J.; Melchionna, T.; Chillakuri, C.; et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat. Immunol. 2012, 13, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvon, E.S.; Vigouroux, S.; Rousseau, R.F.; Biagi, E.; Amrolia, P.; Dotti, G.; Wagner, H.J.; Brenner, M.K. Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood 2003, 102, 3815–3821. [Google Scholar] [CrossRef]
- Del Álamo, D.; Rouault, H.; Schweisguth, F. Mechanism and significance of cis-inhibition in Notch signalling. Curr. Biol. 2011, 21, R40–R47. [Google Scholar] [CrossRef]
- Jeffery, P.K.; Wardlaw, A.J.; Nelson, F.C.; Collins, J.V.; Kay, A.B. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am. Rev. Respir. Dis. 1989, 140, 1745–1753. [Google Scholar] [CrossRef]
- Chiappara, G.; Gagliardo, R.; Siena, A.; Bonsignore, M.R.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Airway remodelling in the pathogenesis of asthma. Curr. Opin. Allergy Clin. Immunol. 2001, 1, 85–93. [Google Scholar] [CrossRef]
- Phipps, S.; Benyahia, F.; Ou, T.T.; Barkans, J.; Robinson, D.S.; Kay, A.B. Acute allergen-induced airway remodeling in atopic asthma. Am. J. Respir. Mol. Biol. 2004, 31, 626–632. [Google Scholar] [CrossRef]
- Leigh, R.; Ellis, R.; Wattie, J.; Southam, D.S.; de Hoogh, M.; Gauldie, J.; O’Byrne, P.M.; Inman, M.D. Dysfunction and remodeling of the mouse airway persist after resolution of acute allergen-induced airway inflammation. Am. J. Respir. Mol. Biol. 2002, 27, 526–535. [Google Scholar] [CrossRef]
- Kariyawasam, H.H.; Aizen, M.; Barkans, J.; Robinson, D.S.; Kay, A.B. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after alergen challenge in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Leigh, R.; Ellis, R.; Wattie, J.N.; Hirota, J.A.; Matthaei, K.I.; Foster, P.S.; O’Byrne, P.M.; Inman, M.D. Type 2 cytokines in the pathogenesis of sustained airway dysfunction and airway remodeling in mice. Am. J. Respir. Crit. Care Med. 2004, 169, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.G. Airway wall remodelling and hyperresponsiveness: Modelling remodelling in vitro and in vivo. Pulm. Pharmacol. Therap. 2001, 14, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.B.; Phipps, S.; Robinson, D.S. A role for eosinophils in airway remodelling in asthma. Trends Immunol. 2004, 25, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Tsai, M.; Tam, S.Y.; Jones, C.; Zehnder, J.; Galli, S.J. Mast cells can promote the development of multiple features of chronic asthma in mice. J. Clin. Investig. 2006, 116, 1633–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood-Page, P.; Menzies-Gow, A.; Phipps, S.; Ying, S.; Wangoo, A.; Ludwig, M.S.; Barnes, N.; Robinson, D.; Kay, A.B. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Investig. 2003, 112, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormanen, K.R.; Uller, L.; Persson, C.G.A.; Erjefalt, J.S. Allergen exposure of mouse airways evokes remodeling of both bronchi and large pulmonary vessels. Am. J. Respir. Crit. Care Med. 2005, 171, 19–25. [Google Scholar] [CrossRef]
- Li, X.; Wilson, J.W. Increased vascularity of the bronchial mucosa in mild asthma. Am. J. Respir. Crit. Care Med. 1997, 156, 229–233. [Google Scholar] [CrossRef]
- Hoshino, M.; Takahashi, M.; Aoike, N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 2001, 107, 295–301. [Google Scholar] [CrossRef]
- Hoshino, M.; Takahashi, M.; Takai, Y.; Sim, J.; Aoike, N. Inhaled corticosteroids decrease vascularity of the bronchial mucosa in patients with asthma. Clin. Exp. Allergy 2001, 31, 722–730. [Google Scholar] [CrossRef]
- Vrugt, B.; Wilson, S.; Bron, A.; Holgate, S.T.; Djukanovic, R.; Aalbers, R. Bronchial angiogenesis in severe glucocorticoid-dependent asthma. Eur. Respir. J. 2000, 15, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Link, H.; Baluk, P.; Homer, R.J.; Chapoval, S.; Bhandari, V.; Kang, M.J.; Cohn, L.; Kim, Y.K.; McDonald, D.M.; et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat. Med. 2004, 10, 1095–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kapturczak, M.H.; Wasserfall, C.; Glushakova, O.Y.; Campbell-Thompson, M.; Deshane, J.S.; Joseph, R.; Cruz, P.E.; Hauswirth, W.W.; Madsen, K.M.; et al. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 7251–7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, K.G.; Karsan, A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006, 107, 2223–2233. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.S.; Li, J.L.; Generali, D.; Poulsom, R.; Cranston, D.W.; Harris, A.L. Up-regulation of Delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005, 65, 8690–8697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: Implications for modulating ateriogenesis and angiogenesis. Mol. Cell. Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef]
- Williams, C.K.; Li, J.L.; Murga, M.; Harris, A.L.; Tosato, G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 2006, 107, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, A.G.; Iruela-Arispe, M.L. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc. Res. 2010, 86, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Szade, A.; Grochot-Przeczek, A.; Florczyk, U.; Jozkowicz, A.; Dulak, J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life 2015, 67, 145–159. [Google Scholar] [CrossRef]
- Leslie, J.D.; riza-McNaughton, L.; Bermange, A.L.; McAdow, R.; Johnson, S.L.; Lewis, J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 2007, 134, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrom, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Suchting, S.; Freitas, C.; le Noble, F.; Benedito, R.; Breant, C.; Duarte, A.; Eichmann, A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scehnet, J.S.; Jiang, W.; Kumar, S.R.; Krasnoperov, V.; Borges, C.; Ley, E.J.; Duarte, A.; Gill, P.S. Inhibition of Dll4 mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 2007, 109, 4753–4760. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Chieh Lin, H.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Siekmann, A.F.; Lawson, N.D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007, 445, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Lobov, I.B.; Renard, R.A.; Papadopoulos, N.; Gale, N.W.; Thurston, G.; Yancopoulos, G.D.; Wiegand, S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The Notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch signaling in blood vessels: Who is talking to whom about what? Circ. Res. 2007, 100, 1556–1568. [Google Scholar] [CrossRef]
- Radtke, F.; Wilson, A.; Mancini, S.J.C.; MacDonald, H.R. Notch regulation of lymphocyte development and function. Nat. Immunol. 2004, 5, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.; Dai, Y.S.; Chou, Y.B.; Juan, Y.H.; Wang, C.C.; Chiang, B.L. Regulatory T cells negatively regulate neovasculature of airway remodeling via DLL4-Notch signaling. J. Immunol. 2009, 183, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Pearson, C.I. Angiogenesis in the pathogenesis of inflammatory joint and lung diseases. Arthritis Res. Ther. 2001, 3, 147. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Hoey, T. Anti-DLL4, a cancer therapeutic with multiple mechanisms of action. Vasc. Cell 2011, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.-C.; Fischer, M.M.; Hynes, M.; Wu, J.; Kim, E.; Beviglia, L.; Yeung, V.P.; Song, X.; Kapoun, A.M.; Lewicki, J.; et al. Anti-DLL4 has broad spectrum activity in pancreatic cancer dependent on targeting DLL4-Notch signaling in both tumor and vasculature cells. Clin. Cancer Res. 2012, 18, 5374–5386. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A phase I dose escalation and expansion study of the anticancer stem cell agent Demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Strauch, U.G.; Obermeier, F.; Grunwald, N.; Gurster, S.; Dunger, N.; Schultz, M.; Griese, D.P.; Maehler, M.; Scholmerich, J.; Rath, H.C. Influence of intestinal bacteria on induction of regulatory T cells: Lessons from a transfer model of colitis. Gut 2005, 54, 1546–1552. [Google Scholar] [CrossRef]
- Ostman, S.; Rask, C.; Wold, A.E.; Hultkrantz, S.; Telemo, E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Hosono, A.; Yanagibashi, T.; Kihara-Fujioka, M.; Hachimura, S.; Itoh, K.; Hirayama, K.; Takahashi, K.; Kaminogawa, S. Intestinal commensal bacteria promote T cell hyporesponsiveness and down-regulate the serum antibody responses induced by dietary antigen. Immunol. Lett. 2010, 132, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Volchkov, P.Y.; Chervonsky, A.V.; Rudensky, A.Y. A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J. Exp. Med. 2010, 207, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Shore, S.A.; Cho, Y. Obesity and asthma: Microbiome-metabolome interactions. Am. J. Respir. Cell Mol. Boil. 2016, 54, 609–617. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Delta-Like Ligand (DLL) | Jagged | γ-Secretase Inhibitor (GSI) |
|---|---|---|
| DLL4 was up-regulated on BMDCs after RSV infection. Anti-DLL4 Ab-treated animals developed substantially increased AHR [68]. | c-Kit-mutant mice had blunted Jagged2 expression on DCs and diminished Th2 & Th17 response to HDM allergic airway inflammation [62]. | IL-4-deficient recipients of GSI-treated naive CD4+ T cells developed lower levels of AHR, reduced numbers of eosinophils, and lower Th2 cytokine [52]. |
| Treatment of OVA-sensitized and challenged mice with Delta1-Fc decreased AHR and airway inflammation [53]. | BMDCs pulsed with allergen upregulated the expression of Jagged1; transfer of these BMDCs induced AHR and eosinophilic airway inflammation. In vivo treatment with Jagged1-Fc enhanced AHR and airway inflammation [52]. | GSI-treated CD8+ T cells failed to restore OVA-induced airway inflammation and AHR in OVA-sensitized recipient CD8−/− mice [53]. |
| DLL1 ligation endowed antigen-presenting cell functions on mast cells, with expression of MHC-II and OX40L and ability to promote naive CD4+ T cell proliferation and differentiation into Th2 cells [80]. | LPS-stimulated stromal cells induced upregulated expression of Jagged-1 by DCs, but not DLL4, which induced allergic airway inflammation [61]. | Administration of GSI inhibited asthma phenotypes, including eosinophilic airway inflammation, goblet cell metaplasia, AHR, and serum IgE [51]. |
| DCs upregulated DLL4 expression in response to cockroach allergen. Blocking DLL4 in vivo enhanced allergen-induced AHR, Th2 cytokine production and mucus secretion [67]. | Jagged1 over-expression enhanced basal cell differentiation into secretory cells, but not ciliated cell differentiation [63]. | GSI treatment suppressed Th17 responses and ameliorated the development of OVA-induced allergic asthma [77]. |
| Mouse mast cells adhered to DLL1-expressing stromal cells, suggesting Notch ligands provided an adhesion function, facilitating mast cell recruitment into inflammatory sites [81]. | Jagged1 inhibition mitigated Th2-dominated airway inflammation, whilst DLL4 blockage aggravated the asthma phenotypes due to impaired Treg induction. Adoptive transfer of DLL4-expressing antigen-presenting cells promoted Treg expansion [69]. | |
| Anti-DLL4 Ab-treated mice had decreased Th17 cells and mitigated airway inflammation [78]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.-T.; Chiu, C.-J.; Chiang, B.-L. Multi-Faceted Notch in Allergic Airway Inflammation. Int. J. Mol. Sci. 2019, 20, 3508. https://doi.org/10.3390/ijms20143508
Huang M-T, Chiu C-J, Chiang B-L. Multi-Faceted Notch in Allergic Airway Inflammation. International Journal of Molecular Sciences. 2019; 20(14):3508. https://doi.org/10.3390/ijms20143508
Chicago/Turabian StyleHuang, Miao-Tzu, Chiao-Juno Chiu, and Bor-Luen Chiang. 2019. "Multi-Faceted Notch in Allergic Airway Inflammation" International Journal of Molecular Sciences 20, no. 14: 3508. https://doi.org/10.3390/ijms20143508
APA StyleHuang, M. -T., Chiu, C. -J., & Chiang, B. -L. (2019). Multi-Faceted Notch in Allergic Airway Inflammation. International Journal of Molecular Sciences, 20(14), 3508. https://doi.org/10.3390/ijms20143508