Unravelling the MicroRNA-Mediated Gene Regulation in Developing Pongamia Seeds by High-Throughput Small RNA Profiling

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Deep Sequencing of Small RNA Libraries in Developing Pongamia Seeds
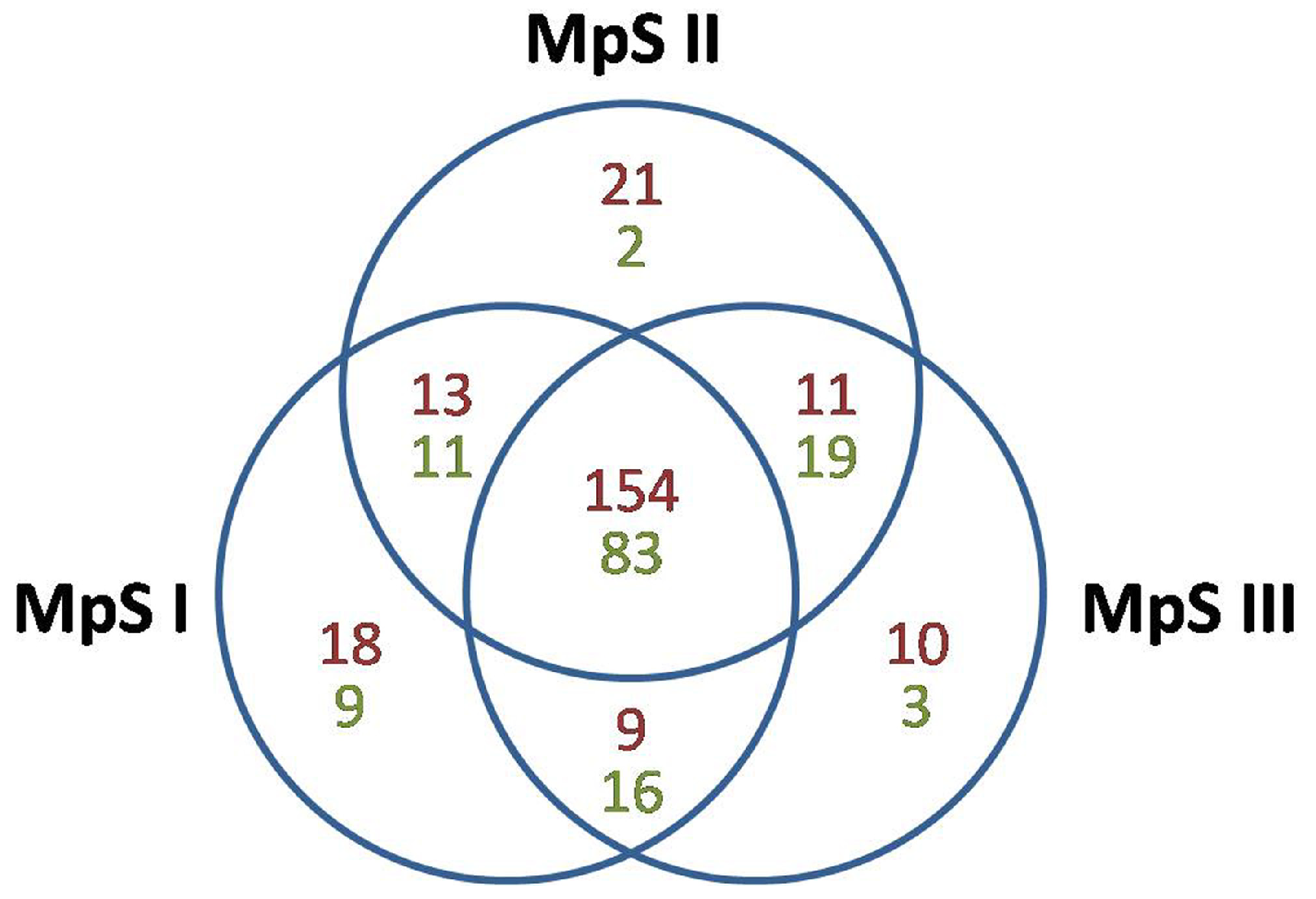
2.2. Identification of Conserved and Novel miRNAs in Developing Pongamia Seeds
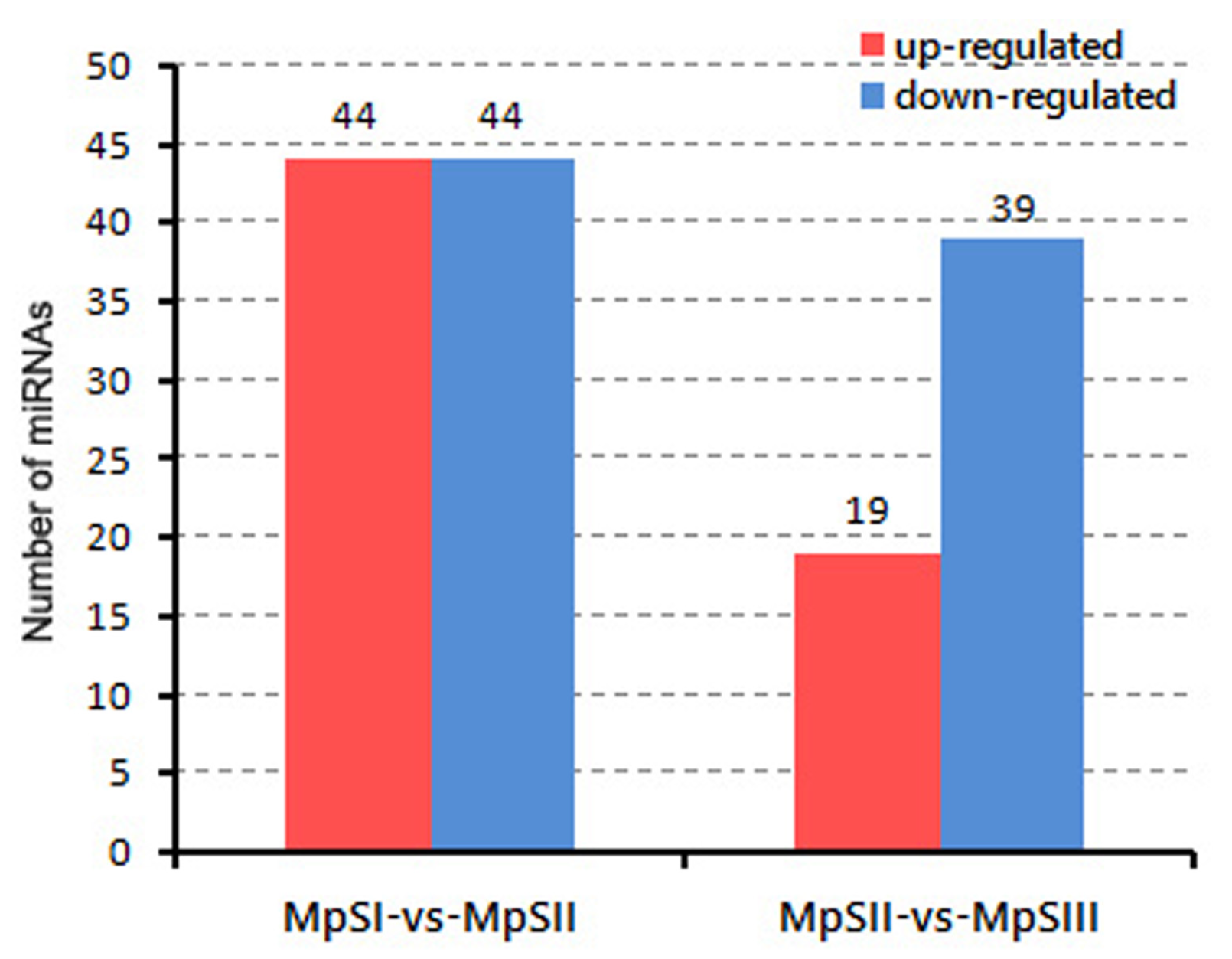
2.3. Differential Expression of miRNAs during Pongamia Seed Development
2.4. Prediction and Functional Annotation of Pongamia miRNA Targets
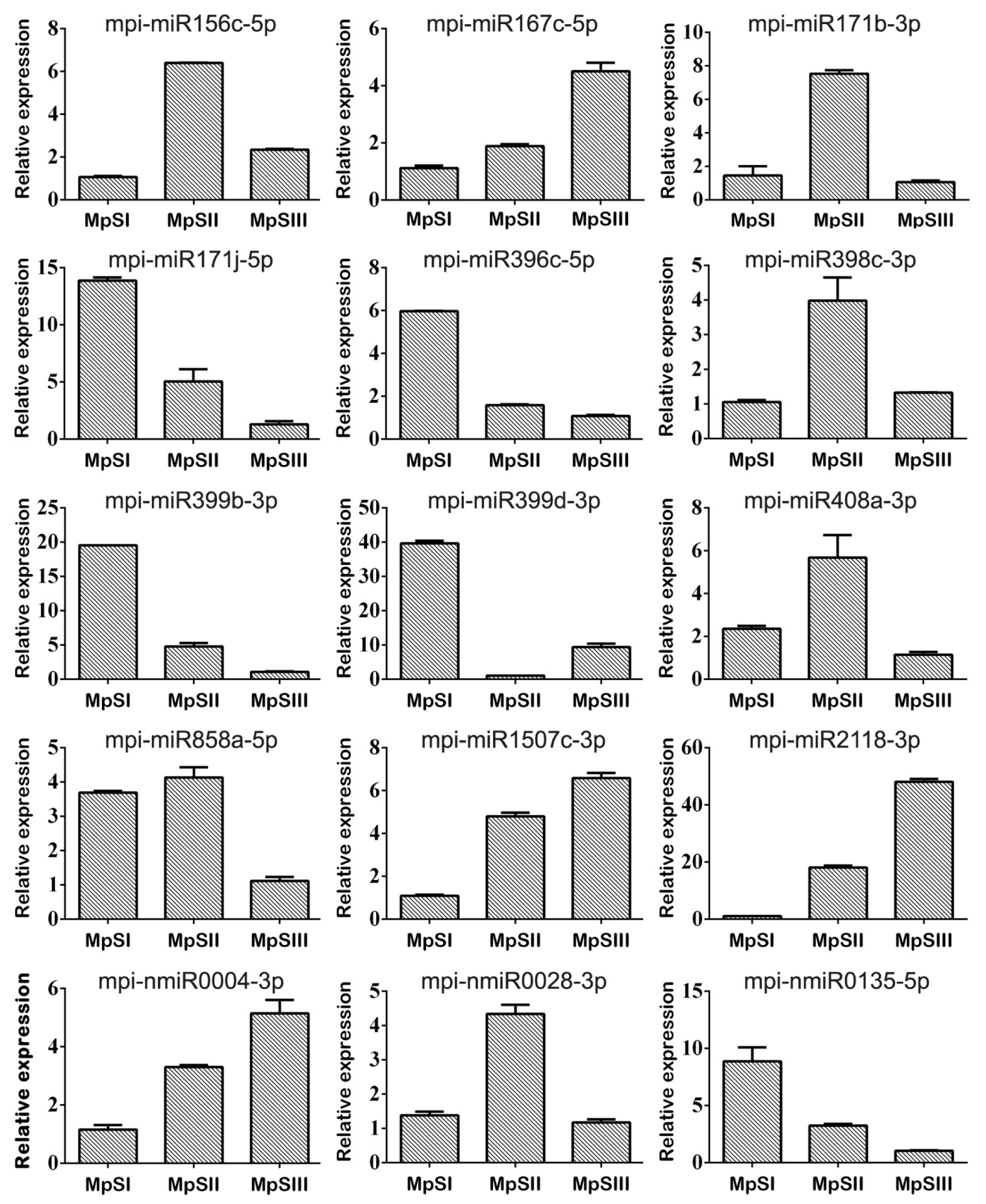
2.5. Validation of DEmiRs during Pongamia Seed Development
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sample Collection
4.2. RNA Isolation, Library Construction and Small RNA Sequencing
4.3. Sequencing Data Processing and Identification of Conserved and Novel miRNAs
4.4. Identification and Analysis of miRNA Targets
4.5. Quantitative Real-Time PCR for miRNAs and Target Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARF | Auxin Response Factor |
| DEmiRs | Differentially Expressed miRNAs |
| NAC | No Apical Meristem |
| qRT-PCR | quantitative Real-Time PCR |
| SPL | SQUAMOSA promoter-binding protein-like |
| TPM | Transcripts Per Million |
| WAF | Weeks After Flowering |
References
- Wang, L.; Yu, H.; He, X.; Liu, R. Influence of fatty acid composition of woody biodiesel plants on the fuel properties. J. Fuel Chem. Technol. 2012, 40, 397–404. [Google Scholar] [CrossRef]
- Balat, M. Potential alternatives to edible oils for biodiesel production-A review of current work. Energy Convers. Manag. 2011, 52, 1479–1492. [Google Scholar] [CrossRef]
- Karmee, S.K.; Chadha, A. Preparation of biodiesel from crude oil of Pongamia pinnata. Bioresour. Technol. 2005, 96, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.; Meher, L.C.; Naik, S.N.; Das, L.M. Production of biodiesel from high free fatty acid Karanja Pongamia pinnata oil. Biomass Bioenerg. 2008, 32, 354–357. [Google Scholar] [CrossRef]
- Sangwan, S.; Rao, D.V.; Sharma, R.A. A review on Pongamia pinnata (L.) Pierre: A great versatile leguminous plant. Nat. Sci. 2010, 8, 130–139. [Google Scholar]
- Huang, J.; Lu, X.; Yan, H.; Chen, S.; Zhang, W.; Huang, R.; Zheng, Y. Transcriptome characterization and sequencing-based identification of salt-responsive genes in Millettia pinnata, a semi-mangrove plant. DNA Res. 2012, 19, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Biswas, B.; Gresshoff, P.M. The role of symbiotic nitrogen fixation in sustainable production of biofuels. Int. J. Mol. Sci. 2014, 15, 7380–7397. [Google Scholar] [CrossRef]
- Gresshoff, P.M.; Hayashi, S.; Biswas, B.; Mirzaei, S.; Indrasumunar, A.; Reid, D.; Samuel, S.; Tollenaere, A.; van Hameren, B.; Hastwell, A.; et al. The value of biodiversity in legume symbiotic nitrogen fixation and nodulation for biofuel and food production. J. Plant Physiol. 2015, 172, 128–136. [Google Scholar] [CrossRef]
- Badole, S.L.; Bodhankar, S.L. Investigation of antihyperglycaemic activity of aqueous and petroleum ether extract of stem bark of Pongamia pinnata on serum glucose level in diabetic mice. J. Ethnopharmacol. 2009, 123, 115–120. [Google Scholar] [CrossRef]
- Al Muqarrabun, L.M.R.; Ahmat, N.; Ruzaina, S.A.S.; Ismail, N.H.; Sahidin, I. Medicinal uses, phytochemistry and pharmacology of Pongamia pinnata (L.) Pierre: A review. J. Ethnopharmacol. 2013, 150, 395–420. [Google Scholar] [CrossRef]
- Roy, R.; Pal, D.; Sur, S.; Mandal, S.; Saha, P.; Panda, C.K. Pongapin and Karanjin, furanoflavanoids of Pongamia pinnata, induce G2/M arrest and apoptosis in cervical cancer cells by differential reactive oxygen species modulation, DNA damage, and nuclear factor kappa-light-chain-enhancer of activated B cell signaling. Phytother. Res. 2019, 33, 1084–1094. [Google Scholar] [PubMed]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.J.; Woody, J.L.; Bolon, Y.; Joseph, B.; Diers, B.W.; Farme, A.D.; Muehlbauer, G.J.; Rex, T.N.; David, G.; James, E.S.; et al. RNA-Seq atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Kong, X.; Chen, F.; Huang, J.; Lou, X.; Zhao, J. Transcriptome analysis of Brassica napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genom. 2015, 16, 858. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, X.; Hao, X.; Zhang, W.; Chen, S.; Huang, R.; Gresshoff, P.M.; Zheng, Y. De novo sequencing and characterization of seed transcriptome of the tree legume Millettia pinnata for gene discovery and SSR marker development. Mol. Breed. 2016, 36, 75. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Whalen, J.; Kinlaw, C.S.; Harry, D.E.; Puryear, J.; Loopstra, C.A.; Gonzalez-Ibeas, D.; Vasquez-Gross, H.A.; Famula, R.A.; Neale, D.B. Transcriptomic profile of leaf tissue from the leguminous tree, Millettia pinnata. Tree Genet. Genomes 2016, 12, 44. [Google Scholar] [CrossRef]
- Sreeharsha, R.V.; Mudalkar, S.; Singha, K.T.; Reddy, A.R. Unravelling molecular mechanisms from floral initiation to lipid biosynthesis in a promising biofuel tree species, Pongamia pinnata using transcriptome analysis. Sci. Rep. 2016, 6, 34315. [Google Scholar] [CrossRef]
- Moolam, A.R.; Singh, A.; Shelke, R.G.; Scott, P.T.; Gresshoff, P.M.; Rangan, L. Identification of two genes encoding microsomal oleate desaturases (FAD2) from the biodiesel plant Pongamia pinnata L. Trees 2016, 30, 1351–1360. [Google Scholar] [CrossRef]
- Ramesh, A.M.; Kesari, V.; Rangan, L. Characterization of a stearoyl-acyl carrier protein desaturase gene from potential biofuel plant, Pongamia pinnata L. Gene 2014, 542, 113–121. [Google Scholar] [CrossRef]
- Winarto, H.P.; Liew, L.C.; Gresshoff, P.M.; Scott, P.T.; Singh, M.B.; Bhalla, P.L. Isolation and characterization of circadian clock genes in the biofuel plant Pongamia (Millettia pinnata). BioEnerg. Res. 2015, 8, 760–774. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Gene Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Korbes, A.P.; Machado, R.; Guzman, F. Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS ONE 2012, 7, e50663. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Wang, M.; Fu, S.X.; Yang, W.C.; Qi, C.K.; Wang, X.J. Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production and development-correlated expression and new small RNA classes. Plant Physiol. 2012, 158, 813–823. [Google Scholar] [CrossRef]
- Goettel, W.; Liu, Z.; Xia, J.; Zhang, W.; Zhao, P.X.; An, Y.Q. Systems and evolutionary characterization of microRNAs and their underlying regulatory networks in soybean cotyledons. PLoS ONE 2014, 9, e86153. [Google Scholar] [CrossRef]
- Song, Q.; Liu, Y.; Hu, X.; Zhang, W.; Ma, B.; Chen, S.; Zhang, J. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 5. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Zhao, K.; Li, F.; Li, K.; Ning, L.; He, J.; Xin, Z.; Yin, D. Small RNA and degradome deep sequencing reveals the roles of microRNAs in seed expansion in peanut Arachis hypogaea L. Front. Plant Sci. 2018, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Xia, H.; Frazier, T.P.; Yao, Y.; Bi, Y.; Li, A.; Li, M.; Li, C.; Zhang, B.; Wang, X. Deep sequencing identifies novel and conserved microRNAs in peanuts Arachis hypogaea L. BMC Plant Biol. 2010, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Galli, V.; Guzman, F.; de Oliveira, L.F.V.; Loss-Morais, G.; Körbes, A.P.; Silva, S.D.A.; Margis-Pinheiro, M.M.A.N.; Margis, R. Identifying microRNAs and transcript targets in Jatropha seeds. PLoS ONE 2014, 9, e83727. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Li, S.; Shu, Q.; Gu, Z.; Wu, Q.; Feng, C.; Xu, W.; Wang, L. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene 2018, 666, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hao, X.; Jin, Y.; Guo, X.; Shao, Q.; Kumar, K.S.; Ahlawat, Y.K.; Harry, D.E.; Joshi, C.P.; Zheng, Y. Temporal transcriptome profiling of developing seeds reveals a concerted gene regulation in relation to oil accumulation in Pongamia (Millettia pinnata). BMC Plant Biol. 2018, 18, 140. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Gene. Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, L.; Yu, M.; Wang, J. Identification and expression analysis of microRNAs during ovule development in rice Oryza sativa by deep sequencing. Plant Cell Rep. 2017, 36, 1815–1827. [Google Scholar] [CrossRef]
- Szittya, G.; Moxon, S.; Santos, D.M.; Jing, R.; Fevereiro, M.P.S.; Moulton, V.; Dalmay, T. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genom. 2008, 9, 593. [Google Scholar] [CrossRef]
- Zabala, G.; Campos, E.; Varala, K.K.; Bloomfield, S.; Jones, S.I.; Win, H.; Tuteja, J.H.; Calla, B.; Clough, S.J.; Hudson, M.; et al. Divergent patterns of endogenous small RNA populations from seed and vegetative tissues of Glycine max. BMC Plant Biol. 2012, 12, 177. [Google Scholar] [CrossRef]
- Fahlgren, N.; Jogdeo, S.; Kasschau, K.D.; Sullivan, C.M.; Chapman, E.J.; Laubinger, S.; Smith, L.M.; Dasenko, M.; Givan, S.A.; Weigel, D.; et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 2010, 22, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.S.; Islam, M.A.; Negi, M.S.; Tripathi, S.B. Changes in oil content and fatty acid profiles during seed development in Pongamia pinnata(L.) Pierre. Indian J. Plant Physiol. 2015, 20, 281–284. [Google Scholar] [CrossRef]
- Diaz, M.; Pecinkova, P.; Nowicka, A.; Baroux, C.; Sakamoto, T.; Gandha, P.Y.; Jeřábková, H.; Matsunaga, S.; Grossniklaus, U.; Pecinka, A. The SMC5/6 complex subunit NSE4A is involved in DNA damage repair and seed development. Plant Cell 2019, 31, 1579–1597. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Miao, L.; He, J.; Zhang, K.; Li, Y.; Gai, J. Dynamic transcriptome changes related to oil accumulation in developing soybean seeds. Int. J. Mol. Sci. 2019, 20, 2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Li, L. Identification of microRNAs actively involved in fatty acid biosynthesis in developing Brassica napus seeds using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef]
- Reyes, J.L.; Chua, N. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Li, J.; Stahle, M.I.; Dubroué, A.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [PubMed]
- Ohto, M.; Fischer, R.L.; Goldberg, R.B.; Nakamura, K.; Harada, J.J. Control of seed mass by APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3123–3128. [Google Scholar] [CrossRef]
- Ohto, M.; Floyd, S.K.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. Effects of APETALA2 on embryo, endosperm, and seed coat development determine seed size in Arabidopsis. Sex. Plant Reprod. 2009, 22, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-Like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Wei, S.; Bradford, K.J. DELAY OF GERMINATION1 (DOG1) Regulates both seed dormancy and flowering time through microRNA pathways. Proc. Natl. Acad. Sci. USA 2016, 113, E2199–E2206. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.N.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Deng, S.; Su, G.; Zeng, Q.; Shi, S. Isolating high-quality RNA from mangroves without liquid nitrogen. Plant Mol. Biol. Rep. 2004, 22, 197. [Google Scholar] [CrossRef]
- Varkonyi-Gasic, E.; Wu, R.; Marion, W.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Statistical Items | MpSI-1 | MpSI-2 | MpSI-3 | MpSII-1 | MpSII-2 | MpSII-3 | MpSIII-1 | MpSIII-2 | MpSIII-3 |
|---|---|---|---|---|---|---|---|---|---|
| Number of raw reads | 31,711,290 | 31,959,711 | 31,763,941 | 27,733,326 | 33,988,872 | 31,143,787 | 31,167,480 | 32,681,945 | 31,100,220 |
| Number of clean reads | 21,400,622 | 21,401,693 | 21,217,533 | 17,612,476 | 21,414,373 | 19,377,920 | 20,039,686 | 20,872,904 | 19,792,612 |
| Retention rate | 67.49% | 66.96% | 66.80% | 63.51% | 63.00% | 62.22% | 64.30% | 63.87% | 63.64% |
| Number of unique tags | 2,757,270 | 2,876,894 | 2,882,107 | 3,066,337 | 3,427,062 | 2,942,567 | 2,593,393 | 2,774,500 | 2,443,701 |
| Number of conserved miRNAs | 174 | 172 | 168 | 157 | 165 | 183 | 163 | 162 | 160 |
| Number of novel miRNAs | 87 | 82 | 68 | 70 | 64 | 83 | 97 | 93 | 94 |
| miRNA_ID | Reads Count | Fold Change | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MpSI-1 | MpSI-2 | MpSI-3 | MpSII-1 | MpSII-2 | MpSII-3 | MpSIII-1 | MpSIII-2 | MpSIII-3 | MpSII/MpSI | MpSIII/MpSII | |
| mpi-miR156f-5p | 0 | 0 | 0 | 2 | 2 | 4 | 12 | 13 | 12 | 7.65 | 1.74 |
| mpi-miR482a-3p | 2380 | 4221 | 2288 | 5869 | 5817 | 7435 | 39410 | 19325 | 18703 | 1.58 | 1.49 |
| mpi-nmiR0004-3p | 21 | 20 | 9 | 25 | 22 | 24 | 69 | 80 | 106 | 1.07 | 1.28 |
| mpi-nmiR0083-3p | 0 | 0 | 0 | 11 | 11 | 9 | 71 | 70 | 84 | 9.71 | 2.28 |
| mpi-nmiR0119-5p | 0 | 0 | 0 | 2 | 2 | 6 | 10 | 8 | 14 | 7.92 | 1.26 |
| mpi-miR156a-3p | 2112 | 3098 | 1195 | 736 | 356 | 577 | 316 | 347 | 424 | −1.39 | −1.19 |
| mpi-miR164a-5p | 141 | 121 | 130 | 7 | 20 | 32 | 11 | 7 | 7 | −2.37 | −1.67 |
| mpi-miR166a-5p | 3344 | 2597 | 2102 | 718 | 822 | 1330 | 437 | 495 | 803 | −1.03 | −1.22 |
| mpi-miR166k-5p | 408 | 297 | 290 | 19 | 21 | 30 | 7 | 4 | 9 | −3.37 | −2.32 |
| mpi-miR171j-5p | 66 | 54 | 35 | 5 | 5 | 5 | 0 | 2 | 0 | −2.83 | −3.43 |
| mpi-miR393b-5p | 384 | 272 | 219 | 44 | 84 | 133 | 35 | 38 | 42 | −1.31 | −1.64 |
| mpi-miR399a-3p | 5907 | 4743 | 3838 | 908 | 1026 | 1399 | 725 | 552 | 741 | −1.65 | −1.24 |
| mpi-miR399b-3p | 3876 | 4196 | 2814 | 1094 | 848 | 1048 | 412 | 207 | 306 | −1.35 | −2.25 |
| miRNA_ID | Reads Count | Fold Change | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MpSI-1 | MpSI-2 | MpSI-3 | MpSII-1 | MpSII-2 | MpSII-3 | MpSIII-1 | MpSIII-2 | MpSIII-3 | MpSII/MpSI | MpSIII/MpSII | |
| mpi-miR156c-5p | 84 | 111 | 69 | 327 | 203 | 197 | 95 | 68 | 83 | 2.01 | −2.16 |
| mpi-miR168d-3p | 0 | 0 | 0 | 2 | 4 | 9 | 0 | 0 | 0 | 8.49 | −8.49 |
| mpi-miR171b-3p | 96 | 94 | 52 | 170 | 226 | 432 | 219 | 142 | 198 | 2.22 | −1.02 |
| mpi-miR398c-3p | 393 | 398 | 260 | 964 | 1092 | 1553 | 360 | 317 | 436 | 2.25 | −2.21 |
| mpi-miR398f-3p | 10 | 10 | 6 | 113 | 142 | 169 | 9 | 7 | 13 | 4.52 | −4.40 |
| mpi-miR408a-3p | 130 | 137 | 75 | 379 | 446 | 612 | 176 | 170 | 199 | 2.56 | −1.91 |
| mpi-miR408b-3p | 15 | 15 | 6 | 68 | 98 | 147 | 35 | 45 | 59 | 3.62 | −1.66 |
| mpi-miR408d-3p | 7 | 8 | 8 | 84 | 63 | 76 | 37 | 54 | 45 | 3.75 | −1.28 |
| mpi-miR4403-5p | 3 | 3 | 3 | 6 | 5 | 6 | 2 | 2 | 2 | 1.39 | −2.06 |
| mpi-miR5037 | 0 | 0 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 7.32 | −7.32 |
| mpi-nmiR0028-3p | 7 | 6 | 7 | 25 | 32 | 19 | 7 | 7 | 3 | 2.44 | −2.75 |
| mpi-nmiR0034-3p | 4 | 5 | 3 | 6 | 6 | 9 | 4 | 2 | 3 | 1.28 | −1.73 |
| mpi-nmiR0050-5p | 0 | 0 | 0 | 5 | 3 | 6 | 0 | 0 | 0 | 8.50 | −8.50 |
| mpi-nmiR0072-5p | 14 | 25 | 12 | 16 | 28 | 47 | 2 | 8 | 5 | 1.25 | −3.06 |
| mpi-nmiR0103-5p | 0 | 0 | 0 | 4 | 16 | 10 | 0 | 0 | 0 | 9.63 | −9.63 |
| mpi-miR396c-3p | 1094 | 422 | 725 | 69 | 78 | 151 | 401 | 240 | 257 | −2.50 | 1.13 |
| mpi-miR396e-5p | 9 | 8 | 4 | 0 | 0 | 0 | 5 | 3 | 5 | −8.57 | 7.88 |
| mpi-miR399d-3p | 43 | 33 | 29 | 0 | 0 | 0 | 5 | 7 | 2 | −10.94 | 7.99 |
| miRNA_ID | Target_ID | Target_Annotation | MpSI_TPM | MpSII_TPM | MpSIII_TPM |
|---|---|---|---|---|---|
| mpi-miR168f-5p | Unigene24517 | cardiolipin synthase (CMP-forming), mitochondrial | 0.01 | 0.01 | 0.36 |
| mpi-nmiR0017-3p | Unigene10960 | 3-ketoacyl-CoA synthase 4 | 1.02 | 1.02 | 0.01 |
| Unigene15710 | linoleate 13S-lipoxygenase 3-1, chloroplastic | ||||
| Unigene26514 | phospholipid:diacylglycerol acyltransferase 1-like | ||||
| Unigene49632 | 3-ketoacyl-CoA synthase 4 | ||||
| mpi-nmiR0028-3p | Unigene22800 | stearoyl-ACP 9-desaturase 6, chloroplastic | 11.54 | 20.81 | 3.09 |
| mpi-nmiR0038-5p | Unigene22005 | phospholipase A2 | 1.42 | 0.01 | 0.37 |
| mpi-nmiR0102-5p | Unigene4253 | malonyltransferase | 0.01 | 0.01 | 2.53 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Liu, L.; Hao, X.; Harry, D.E.; Zheng, Y.; Huang, T.; Huang, J. Unravelling the MicroRNA-Mediated Gene Regulation in Developing Pongamia Seeds by High-Throughput Small RNA Profiling. Int. J. Mol. Sci. 2019, 20, 3509. https://doi.org/10.3390/ijms20143509
Jin Y, Liu L, Hao X, Harry DE, Zheng Y, Huang T, Huang J. Unravelling the MicroRNA-Mediated Gene Regulation in Developing Pongamia Seeds by High-Throughput Small RNA Profiling. International Journal of Molecular Sciences. 2019; 20(14):3509. https://doi.org/10.3390/ijms20143509
Chicago/Turabian StyleJin, Ye, Lin Liu, Xuehong Hao, David E. Harry, Yizhi Zheng, Tengbo Huang, and Jianzi Huang. 2019. "Unravelling the MicroRNA-Mediated Gene Regulation in Developing Pongamia Seeds by High-Throughput Small RNA Profiling" International Journal of Molecular Sciences 20, no. 14: 3509. https://doi.org/10.3390/ijms20143509
APA StyleJin, Y., Liu, L., Hao, X., Harry, D. E., Zheng, Y., Huang, T., & Huang, J. (2019). Unravelling the MicroRNA-Mediated Gene Regulation in Developing Pongamia Seeds by High-Throughput Small RNA Profiling. International Journal of Molecular Sciences, 20(14), 3509. https://doi.org/10.3390/ijms20143509