Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function



Abstract
:1. Introduction
2. Results
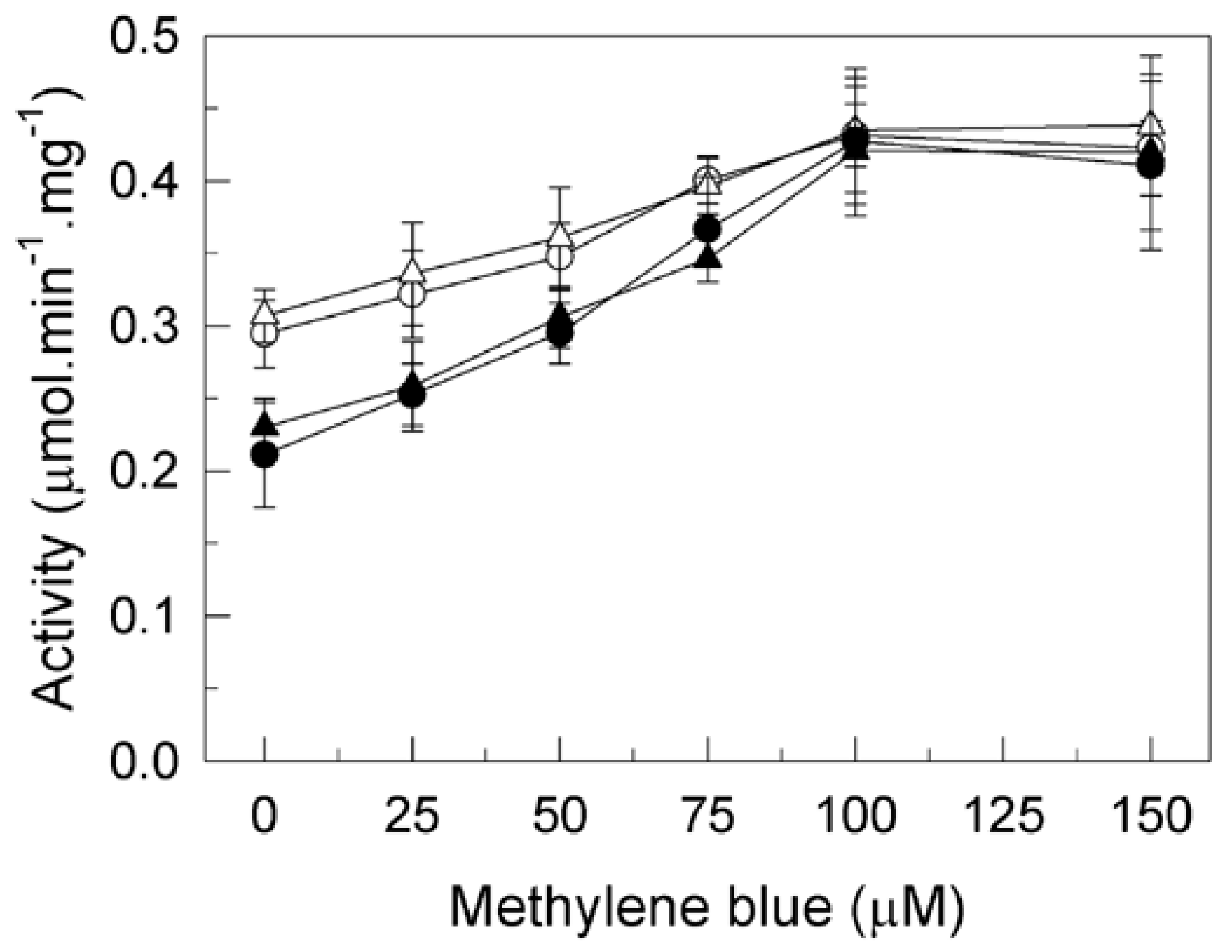
2.1. Methylene Blue (MB) Completely Reverses the Inhibitory Effects of Tau on Plasma Membrane Ca2+-ATPase (PMCA) Activity in Human Brain Membranes of Alzheimer’s Disease Patients and Control Subjects
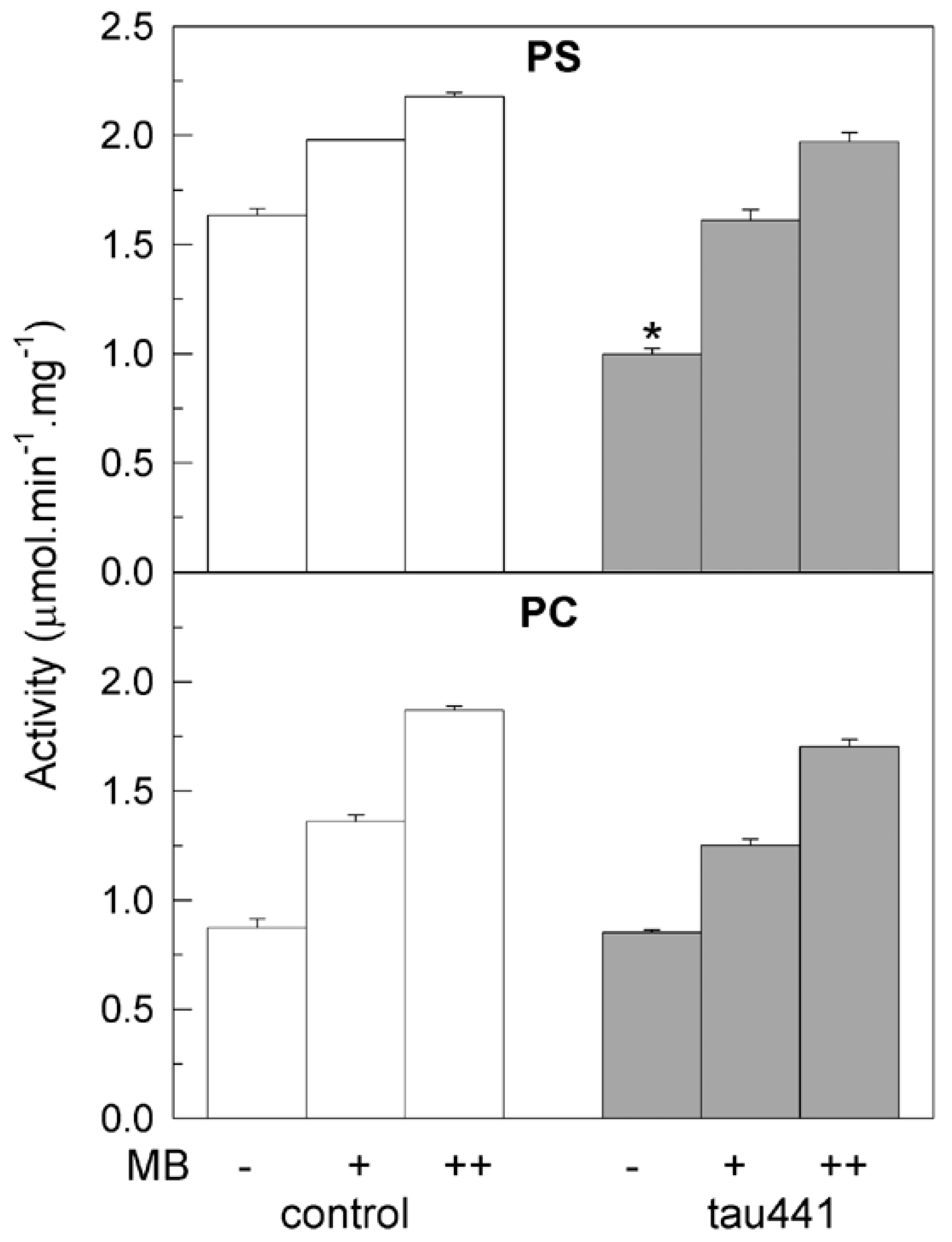
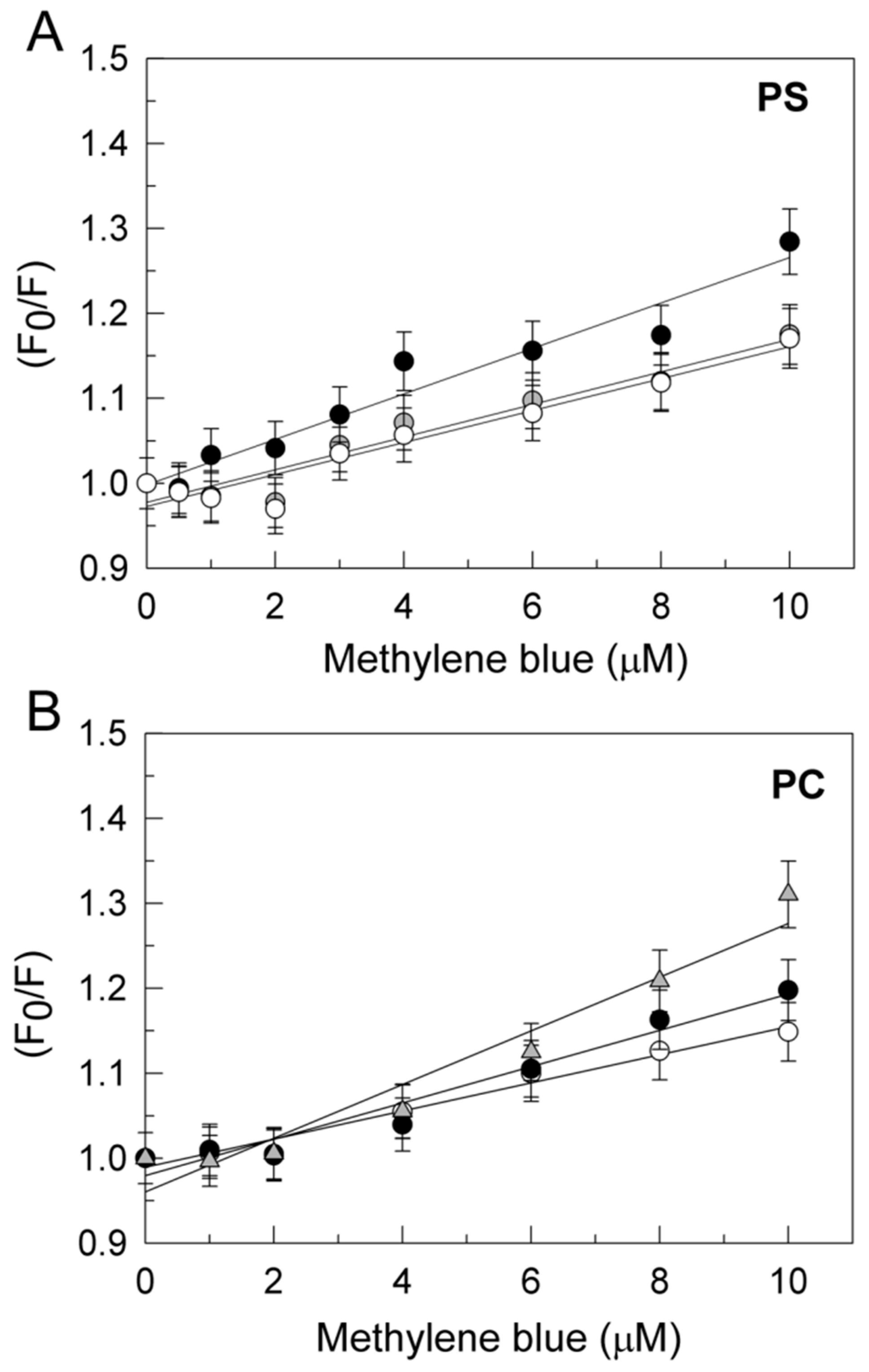
2.2. Methylene Blue Stimulates the Purified PMCA Activity Independently of the Phospholipid Charge and Tau
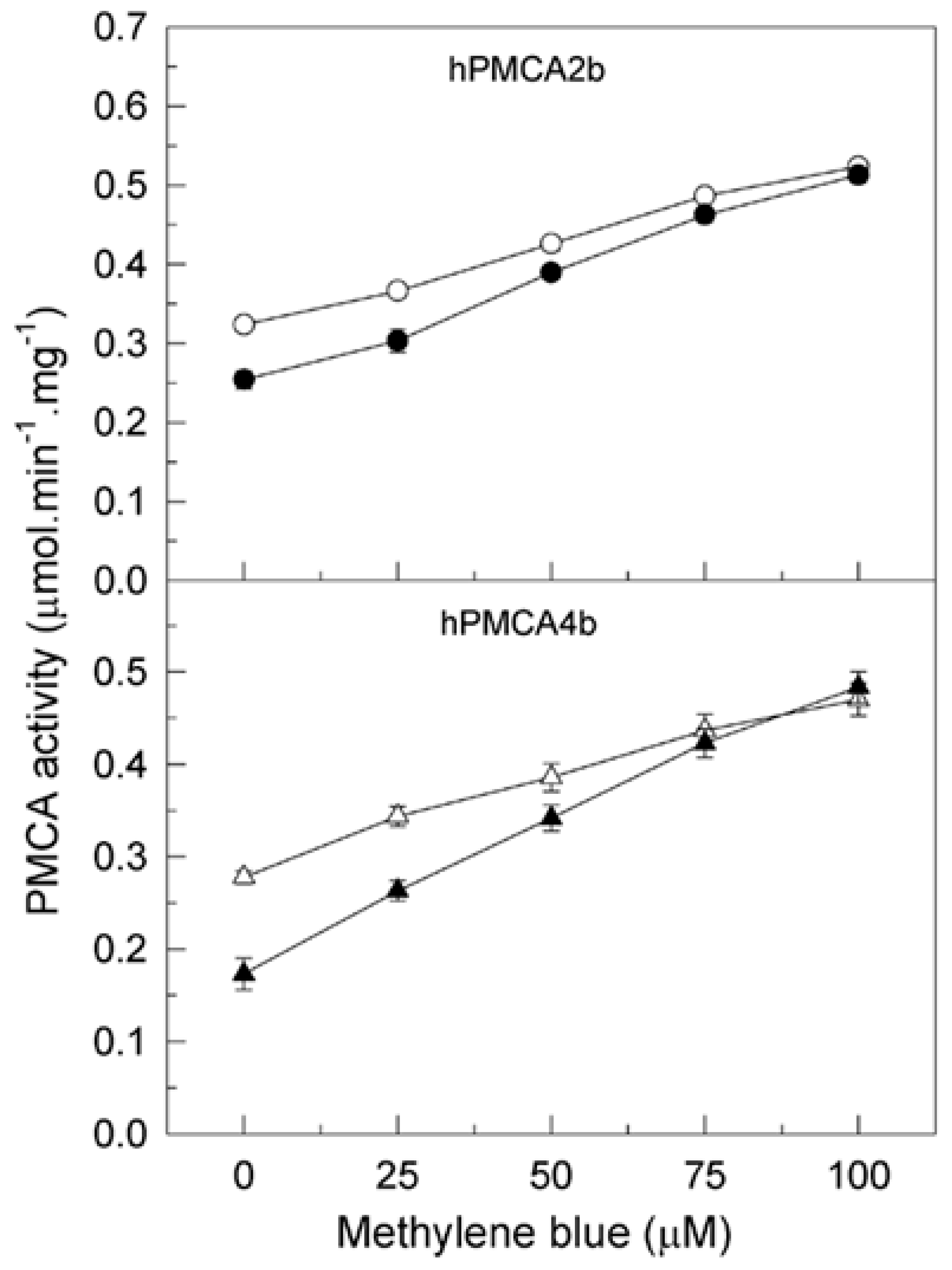
2.3. Methylene Blue Reverses the Inhibitory Effect of Tau on PMCA Isoforms Expressed in COS Cells
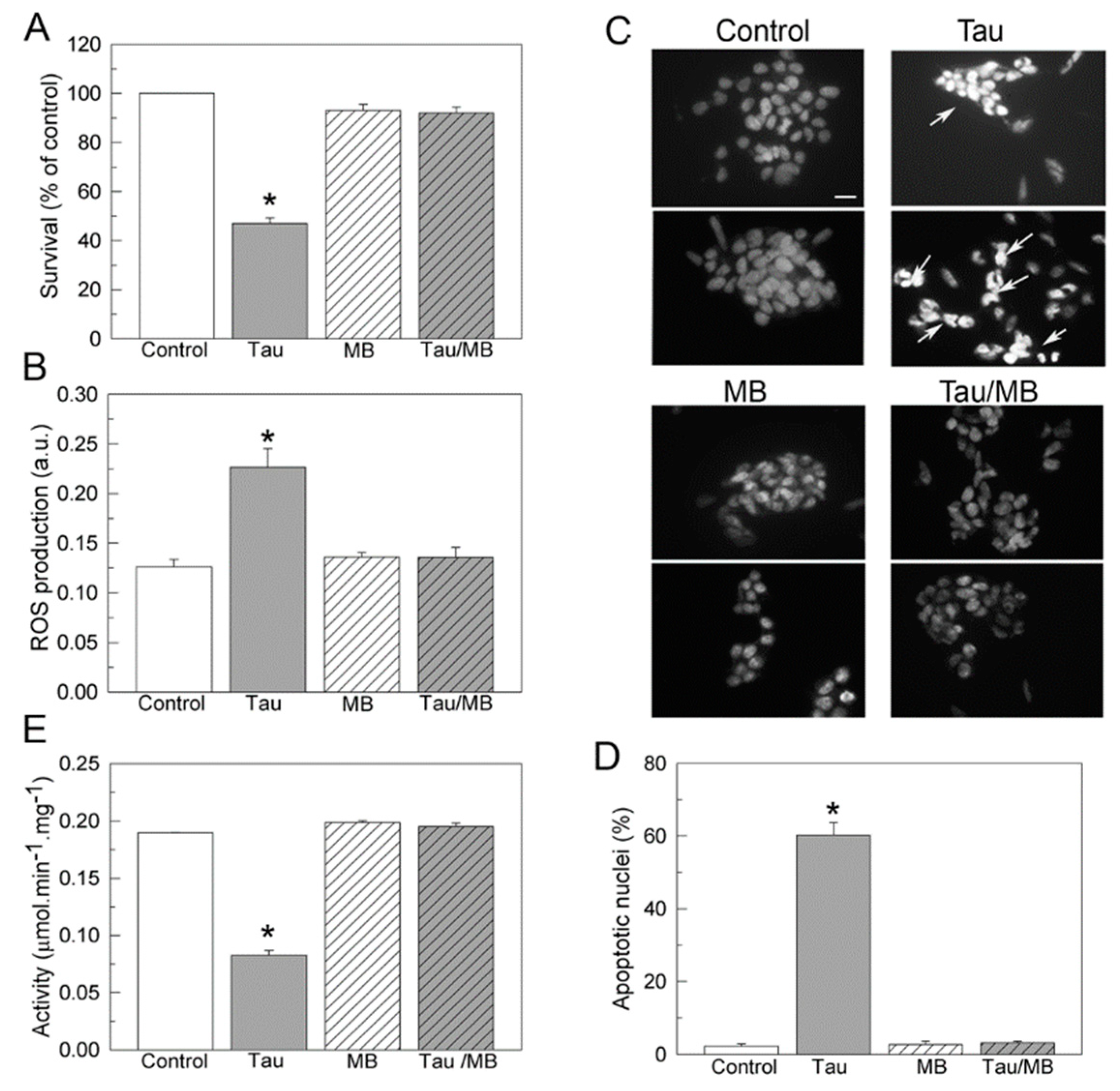
2.4. Methylene Blue Suppresses SH-SY5Y Cells Toxicity Caused by Exogenous Tau and Preserves Endogenous Ca2+-ATPase Activity
2.5. The Interaction of Methylene Blue and PMCA Is Modulated by Tau
3. Discussion
4. Materials and Methods
4.1. Preparation of Membrane Extracts from Cells and Human Brain Tissues
4.2. Preparation of Purified Plasma Membrane Ca2+-ATPase from Pig Brain and Yeast
4.3. Ca2+-ATPase Activity Assays
4.4. Fluorescence Measurements
4.5. Neuroblastoma Cell Cultures and Treatments
4.6. Cell Viability Assay
4.7. Reactive Oxygen Species Assay
4.8. Quantification of Apoptotic Cells
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, G.; Nagbanshi, M.; Goldau, N.; Mendes Jorge, M.; Meissner, P.; Jahn, A.; Mockenhaupt, F.P.; Muller, O. Efficacy and safety of methylene blue in the treatment of malaria: A systematic review. BMC Med. 2018, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Telch, M.J.; Bruchey, A.K.; Rosenfield, D.; Cobb, A.R.; Smits, J.; Pahl, S.; Gonzalez-Lima, F. Effects of post-session administration of methylene blue on fear extinction and contextual memory in adults with claustrophobia. Am. J. Psychiatry 2014, 171, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Delport, A.; Harvey, B.H.; Petzer, A.; Petzer, J.P. Methylene blue and its analogues as antidepressant compounds. Metab. Brain Dis. 2017, 32, 1357–1382. [Google Scholar] [CrossRef] [PubMed]
- Oz, M.; Isaev, D.; Lorke, D.E.; Hasan, M.; Petroianu, G.; Shippenberg, T.S. Methylene blue inhibits function of the 5-HT transporter. Br. J. Pharmacol. 2012, 166, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, C.; Hongwan, D.; Kupfer, A.; Lauterburg, B.H. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharm. 2000, 56, 247–250. [Google Scholar] [CrossRef]
- O’Leary, J.C., III; Li, Q.; Marinec, P.; Blair, L.J.; Congdon, E.E.; Johnson, A.G.; Jinwal, U.K.; Koren, J., III; Jones, J.R.; Kraft, C.; et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol. Neurodegener. 2010, 5, 45. [Google Scholar] [CrossRef]
- Huang, L.; Lu, J.; Cerqueira, B.; Liu, Y.; Jiang, Z.; Duong, T.Q. Chronic oral methylene blue treatment in a rat model of focal cerebral ischemia/reperfusion. Brain Res. 2018, 1678, 322–329. [Google Scholar] [CrossRef]
- Shen, Q.; Du, F.; Huang, S.; Rodriguez, P.; Watts, L.T.; Duong, T.Q. Neuroprotective efficacy of methylene blue in ischemic stroke: An MRI study. PLoS ONE 2013, 8, e79833. [Google Scholar] [CrossRef]
- Cui, Z.Q.; Li, W.L.; Luo, Y.; Yang, J.P.; Qu, Z.Z.; Zhao, W.Q. Methylene Blue Exerts Anticonvulsant and Neuroprotective Effects on Self-Sustaining Status Epilepticus (SSSE) Induced by Prolonged Basolateral Amygdala Stimulation in Wistar Rats. Med. Sci. Monit. 2018, 24, 161–169. [Google Scholar] [CrossRef]
- Rojas, J.C.; John, J.M.; Lee, J.; Gonzalez-Lima, F. Methylene blue provides behavioral and metabolic neuroprotection against optic neuropathy. Neurotox. Res. 2009, 15, 260–273. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: Cohort analysis as modified primary outcome in a phase III clinical trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.S.; Clark, M.E.; Hardy, G.A.; Kraan, D.J.; Biondo, E.; Gonzalez-Lima, F.; Cormack, L.K.; Monfils, M.; Lee, H.J. Daily consumption of methylene blue reduces attentional deficits and dopamine reduction in a 6-OHDA model of Parkinson’s disease. Neuroscience 2017, 359, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Biju, K.C.; Evans, R.C.; Shrestha, K.; Carlisle, D.C.B.; Gelfond, J.; Clark, R.A. Methylene Blue Ameliorates Olfactory Dysfunction and Motor Deficits in a Chronic MPTP/Probenecid Mouse Model of Parkinson’s Disease. Neuroscience 2018, 380, 111–122. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Segawa, T.; Maeda, M.; Maruyama, N.; Kinoshita, N.; Hou, H.; Tan, J.; Town, T. Methylene blue modulates beta-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice. J. Biol. Chem. 2014, 289, 30303–30317. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef]
- Gonzalez-Lima, F.; Auchter, A. Protection against neurodegeneration with low-dose methylene blue and near-infrared light. Front. Cell. Neurosci. 2015, 9, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Lima, F.; Barksdale, B.R.; Rojas, J.C. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem. Pharmacol. 2014, 88, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef]
- Lin, A.-L.; Poteet, E.; Du, F.; Gourav, R.C.; Liu, R.; Wen, Y.; Bresnen, A.; Huang, S.; Fox, P.T.; Yang, S.-H.; et al. Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS ONE 2012, 7, e46585. [Google Scholar] [CrossRef]
- Avila, J.; Llorens-Martin, M.; Pallas-Bazarra, N.; Bolos, M.; Perea, J.R.; Rodriguez-Matellan, A.; Hernandez, F. Cognitive Decline in Neuronal Aging and Alzheimer’s Disease: Role of NMDA Receptors and Associated Proteins. Front. Neurosci. 2017, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Klugmann, M.; Ke, Y.D. Adeno-associated virus-based Alzheimer’s disease mouse models and potential new therapeutic avenues. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Perry, G.; Zhu, X.; Boehm, J. Amyloid Beta and tau proteins as therapeutic targets for Alzheimer’s disease treatment: Rethinking the current strategy. Int. J. Alzheimer’s Dis. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Acosta, Y.; Rodriguez-Cruz, E.N.; Orange, F.; De Jesus-Cortes, H.; Madera, B.; Vaquer-Alicea, J.; Ballester, J.; Guinel, M.J.; Bloom, G.S.; Vega, I.E. EFhd2 is a novel amyloid protein associated with pathological tau in Alzheimer’s disease. J. Neurochem. 2013, 125, 921–931. [Google Scholar] [CrossRef]
- Carafoli, E. Calcium signaling: A tale for all seasons. Proc. Natl. Acad. Sci. USA 2002, 99, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Corbacho, I.; Vazquez-Hernandez, M.; Avila, J.; Sepulveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 2015, 1852, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Marcos, D.; Sepulveda, M.R.; Perez, M.; Avila, J.; Mata, A.M. Altered Ca2+ dependence of synaptosomal plasma membrane Ca2+-ATPase in human brain affected by Alzheimer’s disease. Faseb. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-beta peptides-mediated inhibition of brain plasma membrane Ca(2+)-ATPase. Biochim. Biophys. Acta 2012, 1822, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Corbacho, I.; Sepulveda, M.R.; Gutierrez-Merino, C.; Mata, A.M. Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Corbacho, I.; Gutierrez-Merino, C.; Mata, A.M. Methylene blue activates the PMCA activity and cross-interacts with amyloid beta-peptide, blocking Abeta-mediated PMCA inhibition. Neuropharmacology 2018, 139, 163–172. [Google Scholar] [CrossRef]
- Aeschlimann, C.; Cerny, T.; Kupfer, A. Inhibition of (mono)amine oxidase activity and prevention of ifosfamide encephalopathy by methylene blue. Drug Metab. Dispos. 1996, 24, 1336–1339. [Google Scholar]
- Kristiansen, J.E. Dyes, antipsychotic drugs, and antimicrobial activity. Fragments of a development, with special reference to the influence of Paul Ehrlich. Dan. Med. Bull. 1989, 36, 178–185. [Google Scholar]
- Salvador, J.M.; Mata, A.M. Purification of the synaptosomal plasma membrane (Ca2+ + Mg2+)-ATPase from pig brain. Biochem. J. 1996, 315, 183–187. [Google Scholar] [CrossRef]
- DeMarco, S.J.; Strehler, E.E. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J. Biol. Chem. 2001, 276, 21594–21600. [Google Scholar] [CrossRef]
- Burette, A.C.; Strehler, E.E.; Weinberg, R.J. “Fast” plasma membrane calcium pump PMCA2a concentrates in GABAergic terminals in the adult rat brain. J. Comp. Neurol. 2009, 512, 500–513. [Google Scholar] [CrossRef]
- Brandt, P.; Neve, R.L.; Kammesheidt, A.; Rhoads, R.E.; Vanaman, T.C. Analysis of the tissue-specific distribution of mRNAs encoding the plasma membrane calcium-pumping ATPases and characterization of an alternately spliced form of PMCA4 at the cDNA and genomic levels. J. Biol. Chem. 1992, 267, 4376–4385. [Google Scholar] [PubMed]
- May, J.M.; Qu, Z.C.; Whitesell, R.R. Generation of oxidant stress in cultured endothelial cells by methylene blue: Protective effects of glucose and ascorbic acid. Biochem. Pharmacol. 2003, 66, 777–784. [Google Scholar] [CrossRef]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauters, M.; Wattiez, R.; Ris, L. Internalization of the Extracellular Full-Length Tau Inside Neuro2A and Cortical Cells Is Enhanced by Phosphorylation. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Nicholls, S.; Takeda, S.; Fan, Z.; Hyman, B.T. Formation, release, and internalization of stable tau oligomers in cells. J. Neurochem. 2016, 139, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Lopez, E.; Diez-Ballesteros, J.C.; Avila, J.; Hernandez, F.; Bolos, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbacho, I.; Berrocal, M.; Torok, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid beta-peptide to calmodulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E.; Cali, T. The plasma membrane calcium pumps: Focus on the role in (neuro)pathology. Biochem. Biophys. Res. Commun. 2017, 483, 1116–1124. [Google Scholar] [CrossRef]
- Mata, A.M. Functional interplay between plasma membrane Ca(2+)-ATPase, amyloid beta-peptide and tau. Neurosci. Lett. 2018, 663, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Strehler, E.E.; Thayer, S.A. Evidence for a role of plasma membrane calcium pumps in neurodegenerative disease: Recent developments. Neurosci. Lett. 2018, 663, 39–47. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W. Ca(2+) homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 6697–6712. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Avila, J. Our Working Point of View of Tau Protein. J. Alzheimer’s Dis. Jad. 2018, 62, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.Y.; Ege, C.; Winans, A.; Majewski, J.; Wu, G.; Kjaer, K.; Lee, K.Y. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer’s disease amyloid-beta peptide. Proteins 2008, 72, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Jones, E.M.; Dubey, M.; Camp, P.J.; Vernon, B.C.; Biernat, J.; Mandelkow, E.; Majewski, J.; Chi, E.Y. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemistry 2012, 51, 2539–2550. [Google Scholar] [CrossRef]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S.; Ramlall, T.; Rhoades, E. The role of the lipid bilayer in tau aggregation. Biophys. J. 2010, 98, 2722–2730. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement. (New York N.Y.) 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimer’s Dis. Jad. 2015, 44, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Brandt, R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.H.; Kumar, S.; Pinotsi, D.; Tunnacliffe, A.; St George-Hyslop, P.; Mandelkow, E.; Mandelkow, E.M.; Kaminski, C.F.; Kaminski Schierle, G.S. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J. Biol. Chem. 2014, 289, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Pallas-Bazarra, N.; Terreros-Roncal, J.; Perea, J.R.; Jurado-Arjona, J.; Avila, J.; Llorens-Martin, M. Soluble Tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl. Psychiatry 2017, 7, 1267. [Google Scholar] [CrossRef]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Cuadros, R.; Hernandez, F.; Avila, J. Extracellular tau is toxic to neuronal cells. Febs. Lett. 2006, 580, 4842–4850. [Google Scholar] [CrossRef] [Green Version]
- Medina, M.; Avila, J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 2014, 8, 113. [Google Scholar] [CrossRef]
- Diaz-Hernandez, M.; Gomez-Ramos, A.; Rubio, A.; Gomez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 2010, 285, 32539–32548. [Google Scholar] [CrossRef]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Rubio, A.; Diaz-Hernandez, J.I.; Miras-Portugal, M.T.; Avila, J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2009, 19, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, E.; Edwards, A.M.; Aspee, A.; Moran, F.E.; Borsarelli, C.D.; Lissi, E.A.; Gonzalez-Nilo, D.; Poblete, H.; Scaiano, J.C. Photophysics and photochemistry of dyes bound to human serum albumin are determined by the dye localization. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2010, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Xie, Y.; Peng, W.; Peng, Y.K. Measuring the bioactivity and molecular conformation of typically globular proteins with phenothiazine-derived methylene blue in solid and in solution: A comparative study using photochemistry and computational chemistry. J. Photochem. Photobiol. BBiol. 2016, 158, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Surridge, C.D.; Burns, R.G. The difference in the binding of phosphatidylinositol distinguishes MAP2 from MAP2C and Tau. Biochemistry 1994, 33, 8051–8057. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, E.; Titani, K.; Taniguchi, H. Specific binding of acidic phospholipids to microtubule-associated protein MAP1B regulates its interaction with tubulin. J. Biol. Chem. 1997, 272, 22948–22953. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Rapoport, S.I.; Horrocks, L.A. Membrane phospholipid alterations in Alzheimer’s disease: Deficiency of ethanolamine plasmalogens. Neurochem. Res. 1997, 22, 523–527. [Google Scholar] [CrossRef]
- Wells, G.A.; Wilesmith, J.W. The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol. 1995, 5, 91–103. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Mata, A.M. The interaction of ethanol with reconstituted synaptosomal plasma membrane Ca2+ -ATPase. Biochim. Biophys. Acta 2004, 1665, 75–80. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Hidalgo-Sanchez, M.; Mata, A.M. A developmental profile of the levels of calcium pumps in chick cerebellum. J. Neurochem. 2005, 95, 673–683. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Corbacho, I.; Garcia-Prieto, F.F.; Hinojosa, A.E.; Berrocal, M.; Mata, A.M. An improved method for expression and purification of functional human Ca2+ transporter PMCA4b in Saccharomyces cerevisiae. Protein Expr. Purif. 2016, 120, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.R.; Berrocal, M.; Marcos, D.; Wuytack, F.; Mata, A.M. Functional and immunocytochemical evidence for the expression and localization of the secretory pathway Ca2+-ATPase isoform 1 (SPCA1) in cerebellum relative to other Ca2+ pumps. J. Neurochem. 2007, 103, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer US: New York, NY, USA, 2006. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Al-Mousa, F.; Michelangeli, F. Some commonly used brominated flame retardants cause Ca2+-ATPase inhibition, beta-amyloid peptide release and apoptosis in SH-SY5Y neuronal cells. PLoS ONE 2012, 7, e33059. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, H.U.; Abend, M.; Port, M.; Schwerer, M.; Hauck, E.W.; Weidner, W.; Sparwasser, C. Comparative analysis of different apoptosis detection methods in human testicular cancer. Urol. Res. 2004, 32, 332–337. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [Tau] (nM) | KSV (µM−1) | Kd (µM) |
|---|---|---|---|
| BrainPMCA-PS | 0 | 0.019 ± 0.001 | 52.6 ± 2.5 |
| 0.1 | 0.019 ± 0.001 | 52.6 ± 2.5 | |
| 7.5 | 0.027 ± 0.001 | 37.0 ± 1.9 | |
| BrainPMCA-PC | 0 | 0.0165 ± 0.001 | 60.6 ± 3.2 |
| 7.5 | 0.021 ± 0.001 | 47.6 ± 2.3 | |
| 100 | 0.032 ± 0.001 | 31.3 ± 1.5 | |
| hPMCA4b-PS | 0 | 0.014 ± 0.001 | 71.4 ± 3.7 |
| 0.1 | 0.0275 ± 0.001 | 36.4 ± 2.6 | |
| 7.5 | 0.035 ± 0.001 | 28.6 ± 1.3 | |
| hPMCA4b-PC | 0 | 0.019 ± 0.001 | 52.6 ± 2.8 |
| 7.5 | 0.019 ± 0.001 | 52.6 ± 2.7 | |
| 100 | 0.030 ± 0.001 | 33.3 ± 1.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berrocal, M.; Caballero-Bermejo, M.; Gutierrez-Merino, C.; Mata, A.M. Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function. Int. J. Mol. Sci. 2019, 20, 3521. https://doi.org/10.3390/ijms20143521
Berrocal M, Caballero-Bermejo M, Gutierrez-Merino C, Mata AM. Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function. International Journal of Molecular Sciences. 2019; 20(14):3521. https://doi.org/10.3390/ijms20143521
Chicago/Turabian StyleBerrocal, Maria, Montaña Caballero-Bermejo, Carlos Gutierrez-Merino, and Ana M. Mata. 2019. "Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function" International Journal of Molecular Sciences 20, no. 14: 3521. https://doi.org/10.3390/ijms20143521
APA StyleBerrocal, M., Caballero-Bermejo, M., Gutierrez-Merino, C., & Mata, A. M. (2019). Methylene Blue Blocks and Reverses the Inhibitory Effect of Tau on PMCA Function. International Journal of Molecular Sciences, 20(14), 3521. https://doi.org/10.3390/ijms20143521