RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
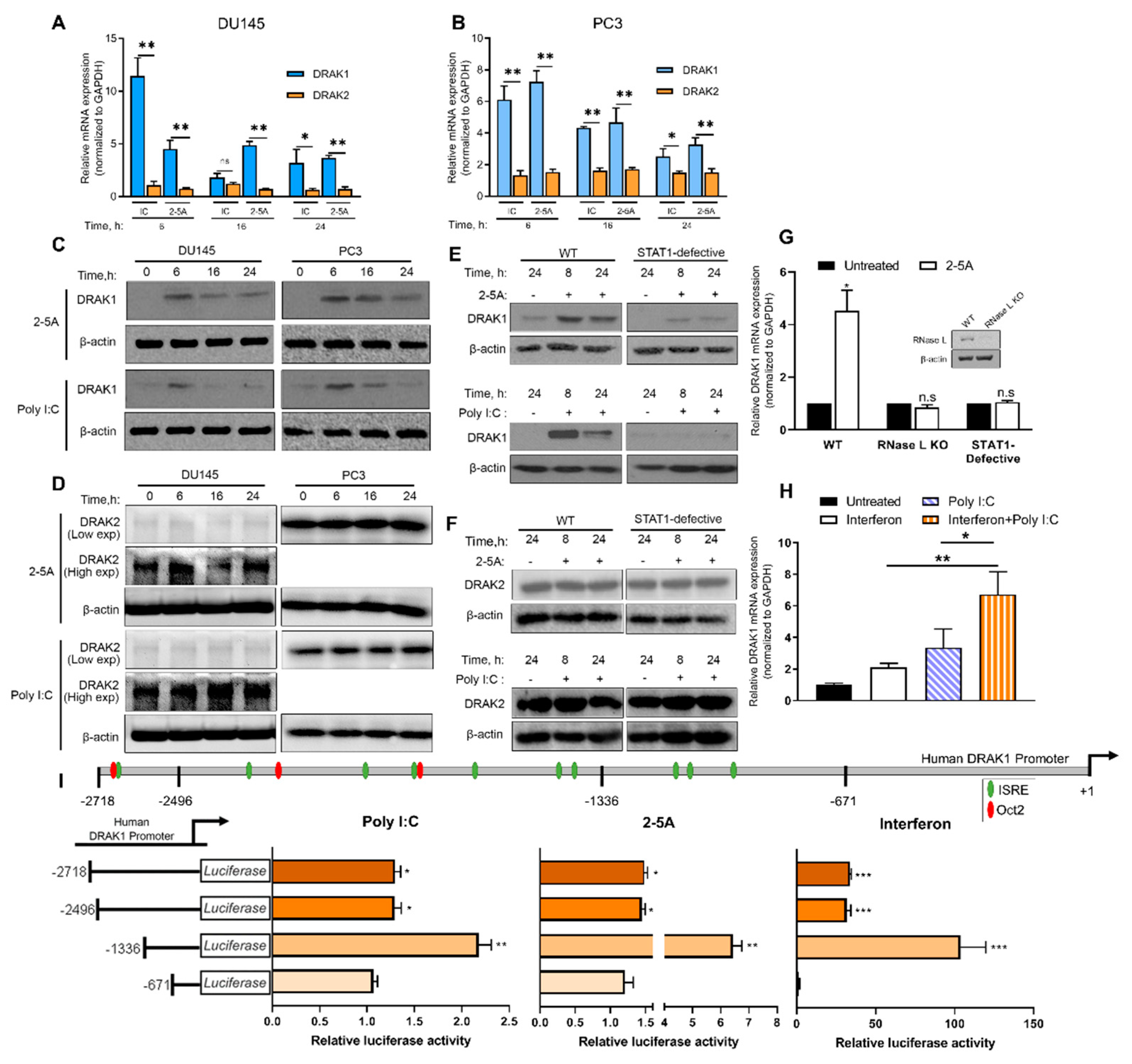
2.1. Induction of DRAK1 by RNase L Requires Interferon Signaling
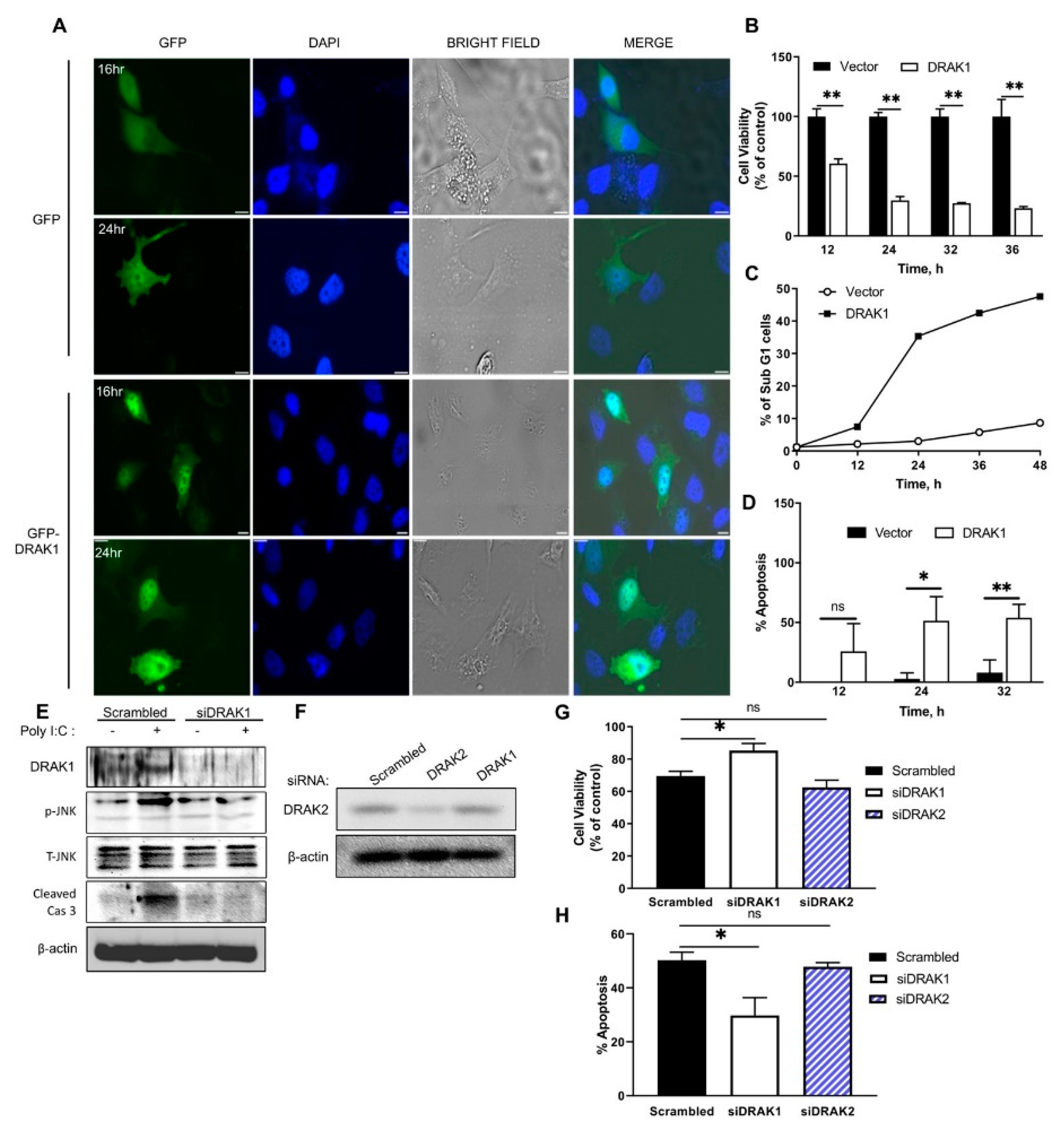
2.2. DRAK1 Localizes to The Nucleus and Induces Apoptosis
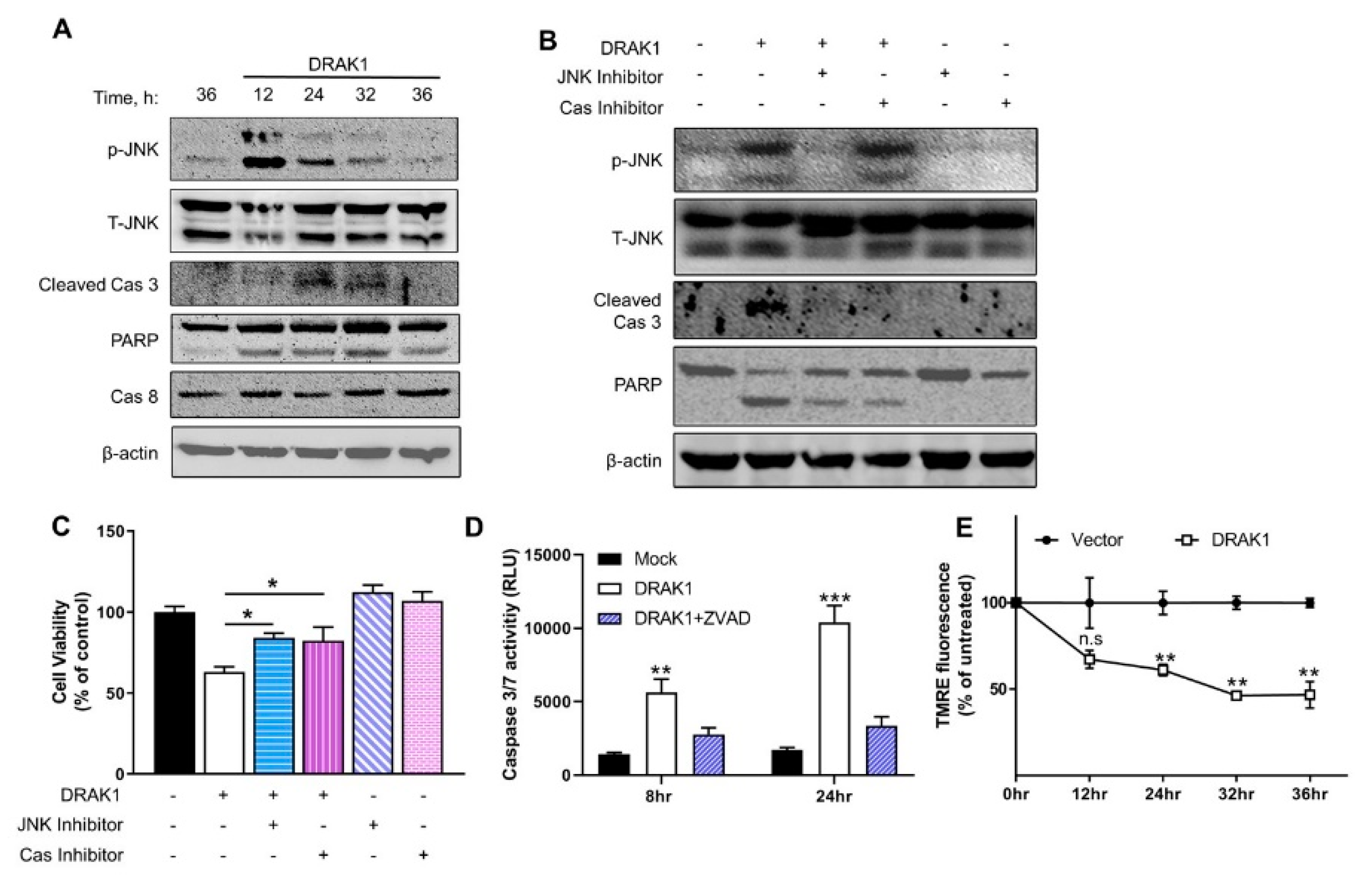
2.3. Ectopic Expression of DRAK1 Induces Apoptosis by Activating JNK and Cleavage of Caspase-3
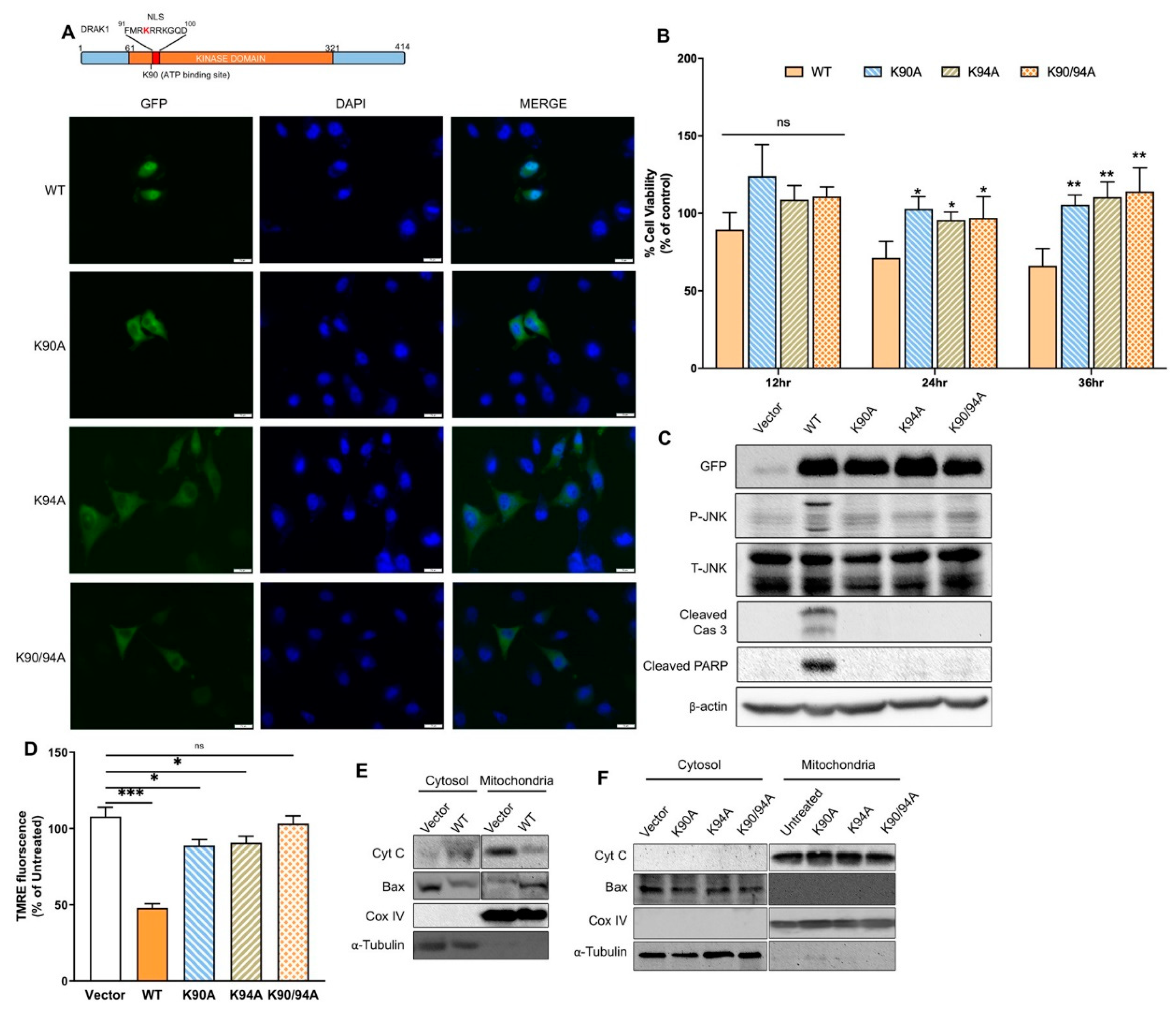
2.4. DRAK1 Kinase Activity and Nuclear Localization Are Required for Inducing Apoptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents and Antibodies
4.2. Cell Culture and Transfections
4.3. Plasmids
4.4. Cell Viability and Caspase 3/7Assays
4.5. Cell Fractionation and Immunoblotting
4.6. RNA Isolation and Quantitative Real Time PCR
- DRAK1-F 5′ TGCTGTGTGAACCTGTCAAAGCAC 3′
- DRAK1-R 5′ACCTGGTTGTCTGAAGTGCCTGAT 3′
- DRAK2-F 5′AAAAATAGGGCATGCGTGTGA 3′
- DRAK2- R 5′ CATAGTTCAGGATTTCTGGAGCTAAA 3′
- GAPDH F 5′ GCAAATTCCATGGCACCGT 3′
- GAPDH R 5′ TCGCCCCACTTGATTTTGG 3′
4.7. Luciferase Reporter Gene Assays
4.8. Tetramethylrhodamine Ethyl Ester (TMRE) Mitochondrial Membrane Potential Assay
4.9. Immunofluorescence Microscopy
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, E.C.; Sen, G.C.; Uzé, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. The interferons and cell death: Guardians of the cell or accomplices of apoptosis? Semin. Cancer Biol. 2000, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Albert, M.L. Dying to Replicate: The Orchestration of the Viral Life Cycle, Cell Death Pathways, and Immunity. Immunity 2011, 35, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, S.M. Viral Subversion of Apoptotic Enzymes: Escape from Death Row. Annu. Rev. Microbiol. 2008, 62, 171–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, S.; Kim, C.N.; Yeh, W.C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef] [PubMed]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef]
- Besch, R.; Poeck, H.; Hohenauer, T.; Senft, D.; Häcker, G.; Berking, C.; Hornung, V.; Endres, S.; Ruzicka, T.; Rothenfusser, S.; et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon–independent apoptosis in human melanoma cells. J. Clin. Investig. 2009, 119, 2399–2411. [Google Scholar] [CrossRef]
- Silverman, R.H. A Scientific Journey Through the 2-5A/RNase L System. Cytokine Growth Factor Rev. 2007, 18, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Paranjape, J.M.; Bulanova, E.; Shim, M.; Guenther-Johnson, J.M.; Faber, P.W.; Eling, T.E.; Williams, B.R.; Silverman, R.H. A transcriptional signaling pathway in the IFN system mediated by 2′-5′-oligoadenylate activation of RNase L. Proc. Natl. Acad. Sci. USA 2005, 102, 14533–14538. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.-M.; Gale, M.; Núñez, G.; Silverman, R.H. RNase L Activates the NLRP3 Inflammasome During Viral Infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Malathi, K. RNase L Induces Autophagy via c-Jun N-terminal Kinase and Double-stranded RNA-dependent Protein Kinase Signaling Pathways. J. Boil. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.A.; Mukherjee, S.; Manivannan, P.; Malathi, K. RNase L Cleavage Products Promote Switch from Autophagy to Apoptosis by Caspase-Mediated Cleavage of Beclin-1. Int. J. Mol. Sci. 2015, 16, 17611–17636. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xiang, Y.; Sabapathy, K.; Silverman, R.H. An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J. Biol. Chem. 2004, 279, 1123–1131. [Google Scholar] [CrossRef]
- Rusch, L.; Zhou, A.; Silverman, R.H. Caspase-Dependent Apoptosis by 2′,5′-Oligoadenylate Activation of RNase L Is Enhanced by IFN-β. J. Interf. Cytokine Res. 2000, 20, 1091–1100. [Google Scholar] [CrossRef]
- Sanjo, H.; Kawai, T.; Akira, S. DRAKs, Novel Serine/Threonine Kinases Related to Death-associated Protein Kinase That Trigger Apoptosis. J. Boil. Chem. 1998, 273, 29066–29071. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Hever, M.P.; Niemaszyk, L.M.; Haghkerdar, J.M.; Yanco, E.G.; Desai, D.; Beyrouthy, M.J.; Kerley-Hamilton, J.S.; Freemantle, S.J.; Spinella, M.J. Serine/Threonine Kinase 17A Is a Novel p53 Target Gene and Modulator of Cisplatin Toxicity and Reactive Oxygen Species in Testicular Cancer Cells. J. Boil. Chem. 2011, 286, 19381–19391. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Hever-Jardine, M.P.; Rahme, G.J.; Yang, E.; Tam, J.; Kodali, A.; Biswal, B.; Fadul, C.E.; Gaur, A.; Israel, M.A.; et al. Serine/Threonine Kinase 17A Is a Novel Candidate for Therapeutic Targeting in Glioblastoma. PLoS ONE 2013, 8, e81803. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.S.; Wardwell-Ozgo, J.; Shah, N.N.; Wright, D.; Appin, C.L.; Vigneswaran, K.; Brat, D.J.; Kornblum, H.I.; Read, R.D. Drak/STK17A Drives Neoplastic Glial Proliferation through Modulation of MRLC Signaling. Cancer Res. 2019, 79, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, W.; Lee, J.M.; Park, J.; Cho, J.K.; Pang, K.; Lee, J.; Kim, D.; Park, S.W.; Yang, K.M.; et al. Cytoplasmic DRAK1 overexpressed in head and neck cancers inhibits TGF-beta1 tumor suppressor activity by binding to Smad3 to interrupt its complex formation with Smad4. Oncogene 2015, 34, 5037–5045. [Google Scholar] [CrossRef] [PubMed]
- Oue, Y.; Murakami, S.; Isshiki, K.; Tsuji, A.; Yuasa, K. Intracellular localization and binding partners of death associated protein kinase-related apoptosis-inducing protein kinase 1. Biochem. Biophys. Res. Commun. 2018, 496, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef] [PubMed]
- Bentrari, F.; Chantome, A.; Knights, A.; Jeannin, J.F.; Pance, A. Oct-2 forms a complex with Oct-1 on the iNOS promoter and represses transcription by interfering with recruitment of RNA PolII by Oct-1. Nucleic Acids Res. 2015, 43, 9757–9765. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K. HPC1/RNASEL Mediates Apoptosis of Prostate Cancer Cells Treated with 2′,5′-Oligoadenylates, Topoisomerase I Inhibitors, and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. Cancer Res. 2004, 64, 9144–9151. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [PubMed] [Green Version]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Inflamm. Process. Mol. Mech. Ther. Oppor. 2000, 103, 13–21. [Google Scholar]
- Dong, C.; Yang, D.D.; Tournier, C.; Whitmarsh, A.J.; Xu, J.; Davis, R.J.; Flavell, R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature 2000, 405, 91–94. [Google Scholar] [CrossRef]
- Hall, J.P.; Merithew, E.; Davis, R.J. c-Jun N-terminal kinase (JNK) repression during the inflammatory response? Just say NO. Proc. Natl. Acad. Sci. USA 2000, 97, 14022–14024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, C. Requirement of JNK for Stress- Induced Activation of the Cytochrome c-Mediated Death Pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Weiss, L.; Whitmarsh, A.J.; Rincón, M.; Davis, R.J.; Flavell, R.A. Regulation of c-Jun NH 2 -terminal Kinase Jnk Gene Expression during T Cell Activation. J. Exp. Med. 2000, 191, 139–146. [Google Scholar]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 Kinase-mediated Phosphorylation of Bax Leads to Its Activation and Mitochondrial Translocation and to Apoptosis of Human Hepatoma HepG2 Cells. J. Boil. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, N.; Becker, E.B.; Konishi, Y.; Bonni, A. JNK Phosphorylation and Activation of BAD Couples the Stress-activated Signaling Pathway to the Cell Death Machinery. J. Boil. Chem. 2002, 277, 40944–40949. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.G.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure-function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef]
- Kuwahara, H.; Nakamura, N.; Kanazawa, H. Nuclear Localization of the Serine/Threonine Kinase DRAK2 Is Involved in UV-Induced Apoptosis. Biol. Pharm. Bull. 2006, 29, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Qiao, X.; Luo, H.; Wu, J. Transgenic Drak2 Overexpression in Mice Leads to Increased T Cell Apoptosis and Compromised Memory T Cell Development. J. Biol. Chem. 2006, 281, 12587–12595. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, H.; Nishizaki, M.; Kanazawa, H. Nuclear localization signal and phosphorylation of Serine350 specify intracellular localization of DRAK2. J. Biochem. 2008, 143, 349–358. [Google Scholar] [CrossRef]
- Friedrich, M.L.; Wen, B.G.; Bain, G.; Kee, B.L.; Katayama, C.; Murre, C.; Hedrick, S.M.; Walsh, C.M. DRAK2, a lymphoid-enriched DAP kinase, regulates the TCR activation threshold during thymocyte selection. Int. Immunol. 2005, 17, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.A.; Scott, D.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manivannan, P.; Reddy, V.; Mukherjee, S.; Clark, K.N.; Malathi, K. RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis. Int. J. Mol. Sci. 2019, 20, 3535. https://doi.org/10.3390/ijms20143535
Manivannan P, Reddy V, Mukherjee S, Clark KN, Malathi K. RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis. International Journal of Molecular Sciences. 2019; 20(14):3535. https://doi.org/10.3390/ijms20143535
Chicago/Turabian StyleManivannan, Praveen, Vidita Reddy, Sushovita Mukherjee, Kirsten Neytania Clark, and Krishnamurthy Malathi. 2019. "RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis" International Journal of Molecular Sciences 20, no. 14: 3535. https://doi.org/10.3390/ijms20143535
APA StyleManivannan, P., Reddy, V., Mukherjee, S., Clark, K. N., & Malathi, K. (2019). RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis. International Journal of Molecular Sciences, 20(14), 3535. https://doi.org/10.3390/ijms20143535