Cardiac Insulin Resistance in Heart Failure: The Role of Mitochondrial Dynamics


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Correlation between HF and Glucose Insufficiency
2.1. Common Coexistence of T2DM in HF Patients
2.2. Antidiabetic Agents and HF Outcome in T2DM Patients
3. Cardiac Metabolic Deficiency under HF
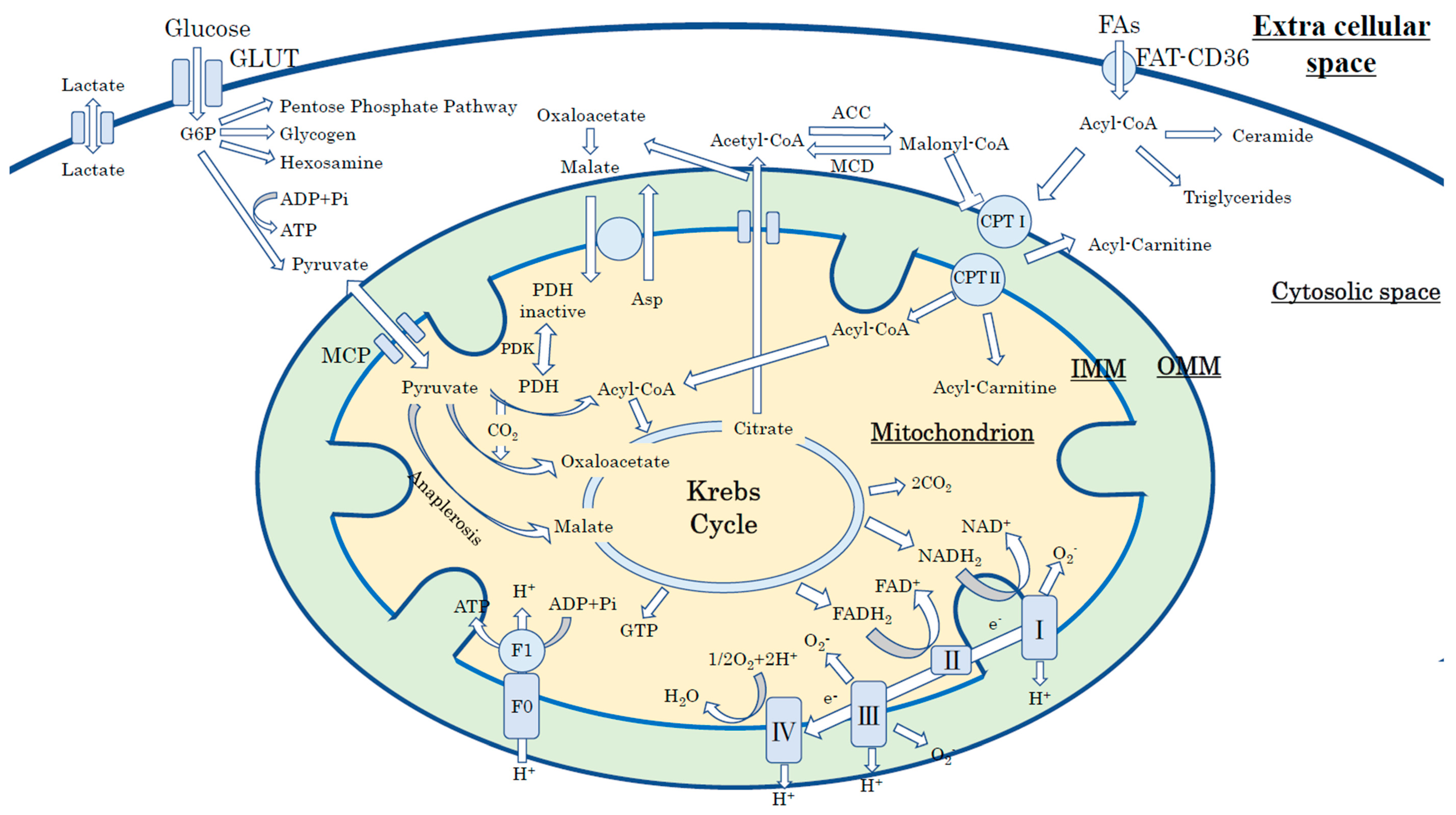
3.1. Cardiac Energy Metabolism in the Physiological Condition
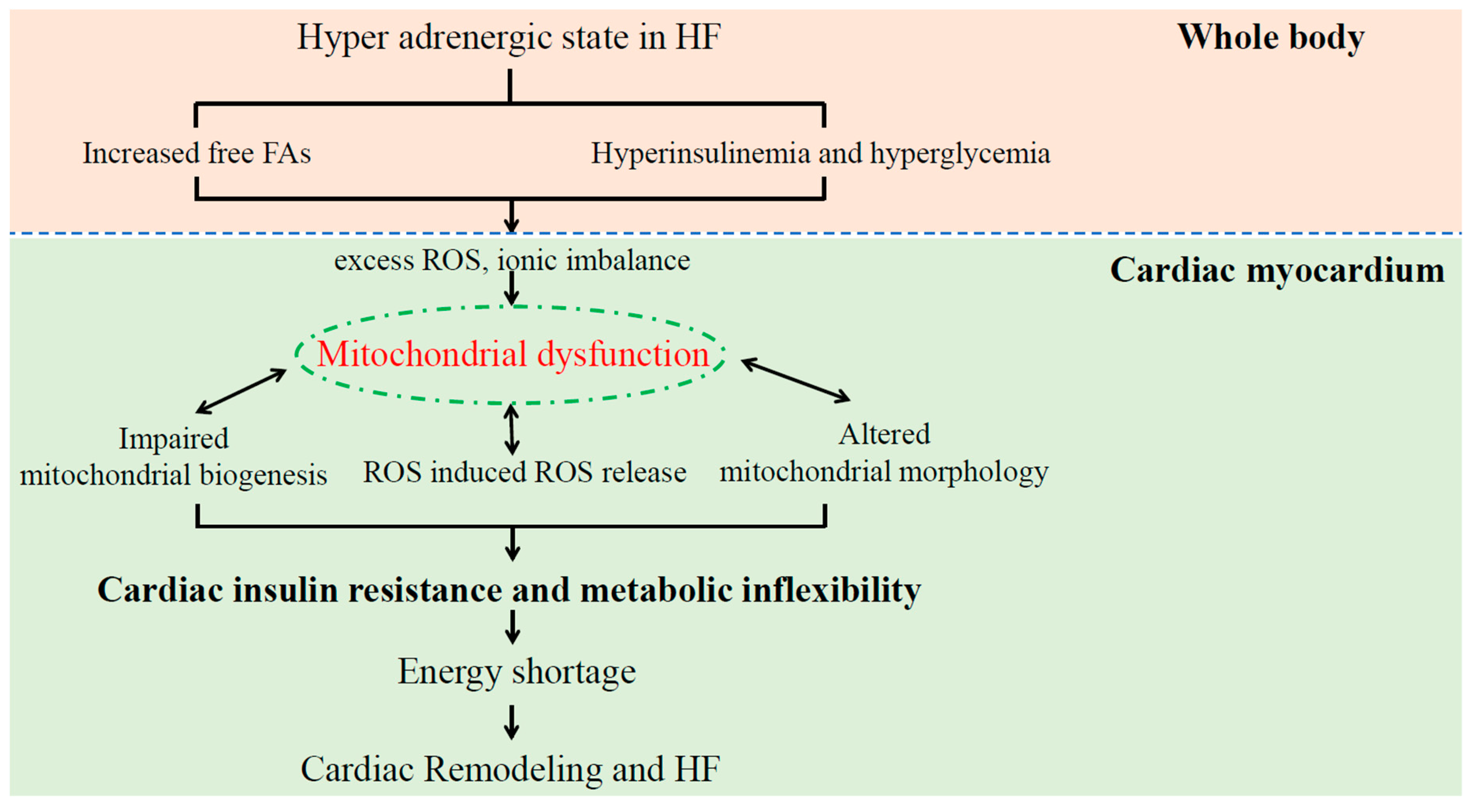
3.2. Cardiac Insulin Resistance and Impaired Cardiac Metabolic Flexibility under HF
3.3. Molecular Mechanism of Impaired Insulin Signaling under HF
4. The Role of Mitochondrial Dynamics in Cardiac Insulin Resistance and HF
4.1. Mitochondrial Energy Production in a Normal Heart
4.2. Mitochondrial Dysfunction Induces Cardiac Insulin Resistance in Patients with HF
4.3. Dysregulation of Mitochondrial Dynamics in HF
5. Cardiac Insulin Resistance in Patients with HF
5.1. Diagnosis of Cardiac Insulin Resistance in Patients with HF and DM
5.2. Innovation of Treatments to Improve Cardiac Insulin Resistance
6. Summary
Funding
Conflicts of Interest
Abbreviations
| T2DM | Type 2 diabetes mellitus |
| HF | Heart failure |
| ATP | Adenosine triphosphate |
| FDG-PET | 18F-2-deoxyglucose positron emission tomography |
| HFrEF | HF with a reduced ejection fraction |
| HFpEF | HF with a preserved ejection fraction |
| HFmrEF | HF with a mid-range ejection fraction |
| mPTP | Mitochondrial permeability transition pore |
| GLUT1/4 | Glucose transporter 1/4 |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| FAs | Fatty acids |
| Mfn1/2 | Mitofusin 1/2 |
| OPA1 | Optic atrophy-1 |
| DRP1 | Dynamin-related protein-1 |
| MFF | Mitochondrial fission factor |
| Fis1 | Mitochondrial fission protein 1 |
| PPAR-χ | Peroxisome proliferator-activated receptor-gamma |
| GLP-1 | Glucagon-like peptide-1 |
| MOMP | Mitochondrial outer membrane permeabilization |
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Udell, J.A.; Cavender, M.A.; Bhatt, D.L.; Chatterjee, S.; Farkouh, M.E.; Scirica, B.M. Glucose-lowering drugs or strategies and cardiovascular outcomes in patients with or at risk for type 2 diabetes: A meta-analysis of randomised controlled trials. Lancet Diabetes Endocrinol. 2015, 3, 356–366. [Google Scholar] [CrossRef]
- Aroor, A.R.; Mandavia, C.H.; Sowers, J.R. Insulin resistance and heart failure: Molecular mechanisms. Heart Fail. Clin. 2012, 8, 609–617. [Google Scholar] [CrossRef]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, C.; Zaugg, M.; Lopaschuk, G.D. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ. Heart Fail. 2013, 6, 1039–1048. [Google Scholar] [CrossRef]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Seferovic, P.M.; Petrie, M.C.; Filippatos, G.S.; Anker, S.D.; Rosano, G.; Bauersachs, J.; Paulus, W.J.; Komajda, M.; Cosentino, F.; de Boer, R.A.; et al. Type 2 diabetes mellitus and heart failure: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 853–872. [Google Scholar] [CrossRef] [PubMed]
- Thrainsdottir, I.S.; Aspelund, T.; Thorgeirsson, G.; Gudnason, V.; Hardarson, T.; Malmberg, K.; Sigurdsson, G.; Ryden, L. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care 2005, 28, 612–616. [Google Scholar] [CrossRef]
- Nichols, G.A.; Hillier, T.A.; Erbey, J.R.; Brown, J.B. Congestive heart failure in type 2 diabetes: Prevalence, incidence, and risk factors. Diabetes Care 2001, 24, 1614–1619. [Google Scholar] [CrossRef]
- Demant, M.N.; Gislason, G.H.; Kober, L.; Vaag, A.; Torp-Pedersen, C.; Andersson, C. Association of heart failure severity with risk of diabetes: A Danish nationwide cohort study. Diabetologia 2014, 57, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Amato, L.; Paolisso, G.; Cacciatore, F.; Ferrara, N.; Ferrara, P.; Canonico, S.; Varricchio, M.; Rengo, F. Congestive heart failure predicts the development of non-insulin-dependent diabetes mellitus in the elderly. The Osservatorio Geriatrico Regione Campania Group. Diabetes Metab. 1997, 23, 213–218. [Google Scholar]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z.; Leor, J.; Freimark, D.; Boyko, V.; Mandelzweig, L.; Adler, Y.; Sherer, Y.; Behar, S. Functional class in patients with heart failure is associated with the development of diabetes. Am. J. Med. 2003, 114, 271–275. [Google Scholar] [CrossRef]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of cardiovascular diseases: A cohort study in 1.9 million people. Lancet Diabetes Endocrinol. 2015, 3, 105–113. [Google Scholar] [CrossRef]
- Pazin-Filho, A.; Kottgen, A.; Bertoni, A.G.; Russell, S.D.; Selvin, E.; Rosamond, W.D.; Coresh, J. HbA 1c as a risk factor for heart failure in persons with diabetes: The Atherosclerosis Risk in Communities (ARIC) study. Diabetologia 2008, 51, 2197–2204. [Google Scholar] [CrossRef]
- Kishimoto, I.; Makino, H.; Ohata, Y.; Tamanaha, T.; Tochiya, M.; Kada, A.; Ishihara, M.; Anzai, T.; Shimizu, W.; Yasuda, S.; et al. Hemoglobin A1c predicts heart failure hospitalization independent of baseline cardiac function or B-type natriuretic peptide level. Diabetes Res. Clin. Pract. 2014, 104, 257–265. [Google Scholar] [CrossRef]
- Suskin, N.; McKelvie, R.S.; Burns, R.J.; Latini, R.; Pericak, D.; Probstfield, J.; Rouleau, J.L.; Sigouin, C.; Solymoss, C.B.; Tsuyuki, R.; et al. Glucose and insulin abnormalities relate to functional capacity in patients with congestive heart failure. Eur. Heart J. 2000, 21, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, S.L.; Preiss, D.; Jhund, P.S.; Squire, I.; Cardoso, J.S.; Merkely, B.; Martinez, F.; Starling, R.C.; Desai, A.S.; Lefkowitz, M.P.; et al. Risk Related to Pre-Diabetes Mellitus and Diabetes Mellitus in Heart Failure With Reduced Ejection Fraction: Insights From Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure Trial. Circ. Heart Fail. 2016, 9, e002560. [Google Scholar] [CrossRef]
- MacDonald, M.R.; Petrie, M.C.; Varyani, F.; Ostergren, J.; Michelson, E.L.; Young, J.B.; Solomon, S.D.; Granger, C.B.; Swedberg, K.; Yusuf, S.; et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: An analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur. Heart J. 2008, 29, 1377–1385. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C.; Swedberg, K.; Carlsson, J.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Pfeffer, M.A.; Yusuf, S.; CHARM Program Investigators. The hemoglobin A1c level as a progressive risk factor for cardiovascular death, hospitalization for heart failure, or death in patients with chronic heart failure: An analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and Morbidity (CHARM) program. Arch. Intern. Med. 2008, 168, 1699–1704. [Google Scholar]
- Dauriz, M.; Targher, G.; Temporelli, P.L.; Lucci, D.; Gonzini, L.; Nicolosi, G.L.; Marchioli, R.; Tognoni, G.; Latini, R.; Cosmi, F.; et al. Prognostic Impact of Diabetes and Prediabetes on Survival Outcomes in Patients With Chronic Heart Failure: A Post-Hoc Analysis of the GISSI-HF (Gruppo Italiano per lo Studio della Sopravvivenza nella Insufficienza Cardiaca-Heart Failure) Trial. J. Am. Heart Assoc. 2017, 6, e005156. [Google Scholar] [CrossRef]
- Eurich, D.T.; Weir, D.L.; Majumdar, S.R.; Tsuyuki, R.T.; Johnson, J.A.; Tjosvold, L.; Vanderloo, S.E.; McAlister, F.A. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: Systematic review of observational studies involving 34,000 patients. Circ. Heart Fail. 2013, 6, 395–402. [Google Scholar] [CrossRef]
- Komajda, M.; McMurray, J.J.; Beck-Nielsen, H.; Gomis, R.; Hanefeld, M.; Pocock, S.J.; Curtis, P.S.; Jones, N.P.; Home, P.D. Heart failure events with rosiglitazone in type 2 diabetes: Data from the RECORD clinical trial. Eur. Heart J. 2010, 31, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Lago, R.M.; Singh, P.P.; Nesto, R.W. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: A meta-analysis of randomised clinical trials. Lancet 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Horita, S.; Nakamura, M.; Satoh, N.; Suzuki, M.; Seki, G. Thiazolidinediones and Edema: Recent Advances in the Pathogenesis of Thiazolidinediones-Induced Renal Sodium Retention. PPAR Res. 2015, 2015, 646423. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.D.; Fonarow, G.C.; Horwich, T.B. Metformin therapy and outcomes in patients with advanced systolic heart failure and diabetes. J. Card. Fail. 2010, 16, 200–206. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm - 2017 Executive Summary. Endocr. Pract. 2017, 23, 207–238. [Google Scholar] [CrossRef]
- MacDonald, M.R.; Petrie, M.C.; Hawkins, N.M.; Petrie, J.R.; Fisher, M.; McKelvie, R.; Aguilar, D.; Krum, H.; McMurray, J.J. Diabetes, left ventricular systolic dysfunction, and chronic heart failure. Eur. Heart J. 2008, 29, 1224–1240. [Google Scholar] [CrossRef]
- Arturi, F.; Succurro, E.; Miceli, S.; Cloro, C.; Ruffo, M.; Maio, R.; Perticone, M.; Sesti, G.; Perticone, F. Liraglutide improves cardiac function in patients with type 2 diabetes and chronic heart failure. Endocrine 2017, 57, 464–473. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. Hemodynamic Effects of Sodium-Glucose Cotransporter 2 Inhibitors. J. Clin. Med. Res. 2017, 9, 457–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Abdurrachim, D.; Luiken, J.J.; Nicolay, K.; Glatz, J.F.; Prompers, J.J.; Nabben, M. Good and bad consequences of altered fatty acid metabolism in heart failure: Evidence from mouse models. Cardiovasc. Res. 2015, 106, 194–205. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Rutter, M.K.; Parise, H.; Benjamin, E.J.; Levy, D.; Larson, M.G.; Meigs, J.B.; Nesto, R.W.; Wilson, P.W.; Vasan, R.S. Impact of glucose intolerance and insulin resistance on cardiac structure and function: Sex-related differences in the Framingham Heart Study. Circulation 2003, 107, 448–454. [Google Scholar] [CrossRef]
- Ingelsson, E.; Sundstrom, J.; Arnlov, J.; Zethelius, B.; Lind, L. Insulin resistance and risk of congestive heart failure. JAMA 2005, 294, 334–341. [Google Scholar] [CrossRef]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac steatosis in diabetes mellitus: A 1H-magnetic resonance spectroscopy study. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef]
- Chandler, M.P.; Kerner, J.; Huang, H.; Vazquez, E.; Reszko, A.; Martini, W.Z.; Hoppel, C.L.; Imai, M.; Rastogi, S.; Sabbah, H.N.; et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1538–H1543. [Google Scholar] [CrossRef]
- O’Donnell, J.M.; Fields, A.D.; Sorokina, N.; Lewandowski, E.D. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J. Mol. Cell. Cardiol. 2008, 44, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, MA, USA, 2001; p. xxiv. 427p. [Google Scholar]
- Saotome, M.; Katoh, H.; Satoh, H.; Nagasaka, S.; Yoshihara, S.; Terada, H.; Hayashi, H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1820–H1828. [Google Scholar] [CrossRef]
- Pacher, P.; Thomas, A.P.; Hajnoczky, G. Ca2+ marks: Miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 2380–2385. [Google Scholar] [CrossRef]
- Eisner, V.; Csordas, G.; Hajnoczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle–pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126 Pt 14, 2965–2978. [Google Scholar] [CrossRef]
- Saotome, M.; Katoh, H.; Yaguchi, Y.; Tanaka, T.; Urushida, T.; Satoh, H.; Hayashi, H. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1125–H1132. [Google Scholar] [CrossRef] [Green Version]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009, 20, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Givvimani, S.; Pushpakumar, S.B.; Metreveli, N.; Veeranki, S.; Kundu, S.; Tyagi, S.C. Role of mitochondrial fission and fusion in cardiomyocyte contractility. Int. J. Cardiol. 2015, 187, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G., II. Mitochondrial fission/fusion and cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 38–44. [Google Scholar] [Green Version]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Rajapurohitam, V.; Kilic, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacin, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Biol. 2009, 41, 1846–1854. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacin, M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch. Physiol. Biochem. 2009, 115, 1–12. [Google Scholar] [CrossRef]
- Boudina, S.; Bugger, H.; Sena, S.; O’Neill, B.T.; Zaha, V.G.; Ilkun, O.; Wright, J.J.; Mazumder, P.K.; Palfreyman, E.; Tidwell, T.J.; et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009, 119, 1272–1283. [Google Scholar] [CrossRef]
- Watanabe, T.; Saotome, M.; Nobuhara, M.; Sakamoto, A.; Urushida, T.; Katoh, H.; Satoh, H.; Funaki, M.; Hayashi, H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp. Cell Res. 2014, 323, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Bonora, E.; Targher, G.; Alberiche, M.; Bonadonna, R.C.; Saggiani, F.; Zenere, M.B.; Monauni, T.; Muggeo, M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: Studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000, 23, 57–63. [Google Scholar] [CrossRef]
- Bacharach, S.L.; Bax, J.J.; Case, J.; Delbeke, D.; Kurdziel, K.A.; Martin, W.H.; Patterson, R.E. PET myocardial glucose metabolism and perfusion imaging: Part 1-Guidelines for data acquisition and patient preparation. J. Nucl. Cardiol. 2003, 10, 543–556. [Google Scholar] [CrossRef]
- Fallavollita, J.A.; Luisi, A.J., Jr.; Yun, E.; deKemp, R.A.; Canty, J.M., Jr. An abbreviated hyperinsulinemic-euglycemic clamp results in similar myocardial glucose utilization in both diabetic and non-diabetic patients with ischemic cardiomyopathy. J. Nucl. Cardiol. 2010, 17, 637–645. [Google Scholar] [CrossRef]
- Knuuti, M.J.; Nuutila, P.; Ruotsalainen, U.; Saraste, M.; Harkonen, R.; Ahonen, A.; Teras, M.; Haaparanta, M.; Wegelius, U.; Haapanen, A.; et al. Euglycemic hyperinsulinemic clamp and oral glucose load in stimulating myocardial glucose utilization during positron emission tomography. J. Nucl. Med. 1992, 33, 1255–1262. [Google Scholar]
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 2010, 31, 100–111. [Google Scholar] [CrossRef]
- Sakamoto, A.; Saotome, M.; Hasan, P.; Satoh, T.; Ohtani, H.; Urushida, T.; Katoh, H.; Satoh, H.; Hayashi, H. Eicosapentaenoic acid ameliorates palmitate-induced lipotoxicity via the AMP kinase/dynamin-related protein-1 signaling pathway in differentiated H9c2 myocytes. Exp. Cell Res. 2017, 351, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Hasan, P.; Saotome, M.; Ikoma, T.; Iguchi, K.; Kawasaki, H.; Iwashita, T.; Hayashi, H.; Maekawa, Y. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J. Mol. Cell. Cardiol. 2018, 121, 103–106. [Google Scholar] [CrossRef]
- Ishikita, A.; Matoba, T.; Ikeda, G.; Koga, J.; Mao, Y.; Nakano, K.; Takeuchi, O.; Sadoshima, J.; Egashira, K. Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart From Ischemia-Reperfusion Injury Through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction. J. Am. Heart Assoc. 2016, 5, e003872. [Google Scholar]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [Green Version]
- Steggall, A.; Mordi, I.R.; Lang, C.C. Targeting Metabolic Modulation and Mitochondrial Dysfunction in the Treatment of Heart Failure. Diseases 2017, 5, 14. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saotome, M.; Ikoma, T.; Hasan, P.; Maekawa, Y. Cardiac Insulin Resistance in Heart Failure: The Role of Mitochondrial Dynamics. Int. J. Mol. Sci. 2019, 20, 3552. https://doi.org/10.3390/ijms20143552
Saotome M, Ikoma T, Hasan P, Maekawa Y. Cardiac Insulin Resistance in Heart Failure: The Role of Mitochondrial Dynamics. International Journal of Molecular Sciences. 2019; 20(14):3552. https://doi.org/10.3390/ijms20143552
Chicago/Turabian StyleSaotome, Masao, Takenori Ikoma, Prottoy Hasan, and Yuichiro Maekawa. 2019. "Cardiac Insulin Resistance in Heart Failure: The Role of Mitochondrial Dynamics" International Journal of Molecular Sciences 20, no. 14: 3552. https://doi.org/10.3390/ijms20143552
APA StyleSaotome, M., Ikoma, T., Hasan, P., & Maekawa, Y. (2019). Cardiac Insulin Resistance in Heart Failure: The Role of Mitochondrial Dynamics. International Journal of Molecular Sciences, 20(14), 3552. https://doi.org/10.3390/ijms20143552