Analysis of Chemically Labile Glycation Adducts in Seed Proteins: Case Study of Methylglyoxal-Derived Hydroimidazolone 1 (MG-H1)

 , , ,
, , , 
 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Protein Isolation and Enzymatic Hydrolysis
2.2. Removal of the Detergent by Solid Phase Extraction
2.3. RP-UHPLC-ESI-LIT-Orbitrap-MS Analysis
2.4. Standardization and Validation of the Quantification Method
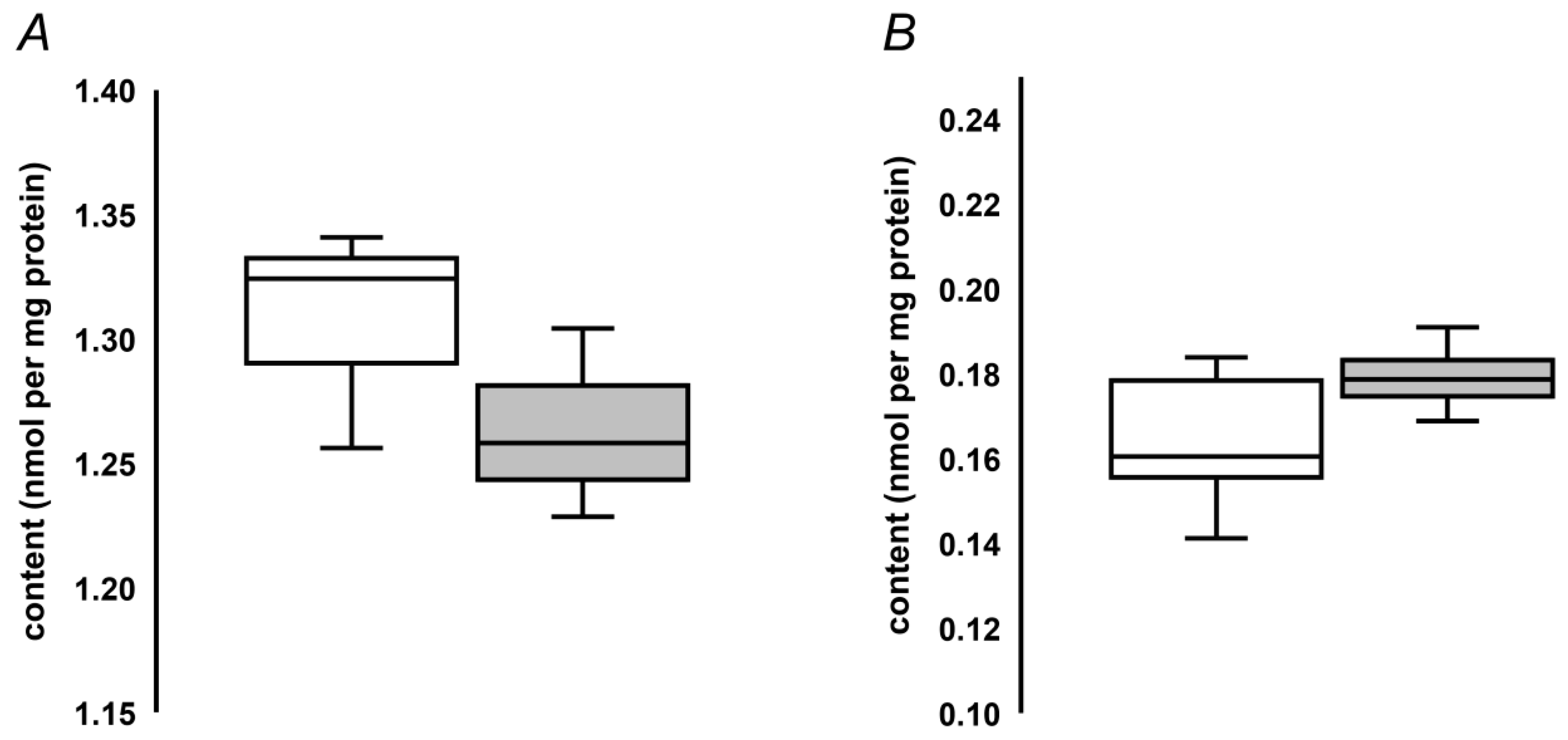
2.5. Quantification of MG-H1 in Seed Protein Hydrolysates by Stable Isotope Dilution
2.6. Compatibility of the Hydrolysis Protocol with Cell Assays
3. Discussion
3.1. Solubilization and Enzymatic Hydrolysis
3.2. Removal of the Detergent from Hydrolysates
3.3. Quantitative Analysis
4. Materials and Methods
4.1. Reagents and Plant Material
4.2. Glycation of Bovine Serum Albumin (BSA)
4.3. Ageing of Pea and Oilseed Rape Seeds
4.4. Protein Isolation
4.5. SDS-PAGE
4.6. Exhaustive Enzymatic Hydrolysis
4.7. Solid Phase Extraction (SPE)
4.8. Derivatization
4.9. RP-UHPLC-ESI-LIT-Orbitrap-MS Analysis
4.10. Method Validation
4.11. Cell Culture
4.12. Analysis of Cell Viability by MTT Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AALS | anionic acid-labile surfactant |
| AGEs | advanced glycation end products |
| AV | average value |
| BSA | bovine serum albumin |
| CEA | Nδ-(carboxyethyl)arginine |
| CMA | Nδ-(carboxymethyl)arginine |
| CML | Nɛ-(carboxymethyl)lysine |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| EDTA | ethylenediaminetetraacetic acid |
| ESI | electrospray ionization |
| FIA-HR-MS | flow injection analysis coupled on-line with high-resolution mass spectrometry |
| HESI | heated electrospray ionization |
| LDR | linear dynamic range |
| L-FDVA | N2-(5-fluoro-2,4-dinitrophenyl)-L-valine amide |
| LOD | limit of detection |
| LOQ | limit of quantification |
| ME | mean error |
| MG-H1 | Nδ-(5-methyl-4-oxo-5-hydroimidazo-linone-2-yl)-L-ornithine, methylglyoxal-derived hydro-imidazolone 1 |
| MG-H2 | 2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid, methylglyoxal-derived hydroimidazolone 2 |
| MG-H3 | 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)pentanoic acid, methylglyoxal-derived hydroimidazolone 3 |
| MGO | methylglyoxal |
| MRM | multiple reaction monitoring |
| MTT | 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | phosphate buffered saline |
| PES | polyethersulfone |
| QqQ | triple quadrupole |
| RAGEs | receptors to advanced glycation end-products |
| RP-UHPLC | reversed phase-ultra-high-performance liquid chromatography |
| RP-SPE | reversed phase-solid phase extraction |
| RSD | relative standard deviation |
| SDS | sodium dodecyl sulfate |
| SDS-PAGE | polyacrylamide gel electrophoresis in sodium dodecyl sulfate |
| SPE | solid phase extraction |
| XIC | extracted ion chromatogram |
References
- Food and Agriculture Organization of the United Nations. The State of Food Security and Nutrition in the World. Building Climate Resilience for Food Security and Nutrition; FAO: Rome, Italy, 2018; p. 108. [Google Scholar]
- Bradford, K.J.; Dahal, P.; van Asbrouck, J.; Kunusoth, K.; Bello, P.; Thompson, J.; Wu, F. The dry chain: Reducing postharvest losses and improving food safety in humid climates. Trends Food Sci. Technol. 2018, 71, 84–93. [Google Scholar] [CrossRef]
- Frolov, A.; Mamontova, T.; Ihling, C.; Lukasheva, E.; Bankin, M.; Chantseva, V.; Vikhnina, M.; Soboleva, A.; Shumilina, J.; Mavropolo-Stolyarenko, G.; et al. Mining seed proteome: From protein dynamics to modification profiles. Biol. Commun. 2018, 63, 43–58. [Google Scholar] [CrossRef]
- Murthy, U.M.N.; Kumar, P.P.; Sun, W.Q. Mechanisms of seed ageing under different storage conditions for Vigna radiata (L.) Wilczek: Lipid peroxidation, sugar hydrolysis, Maillard reactions and their relationship to glass state transition. J. Exp. Bot. 2003, 54, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Hampton, J.G.; TeKrony, D.M. Handbook of Vigour Test. Method; International Seed Testing Association: Zurich, Switzerland, 1995. [Google Scholar]
- Milkovska-Stamenova, S.; Schmidt, R.; Frolov, A.; Birkemeyer, C. GC-MS Method for the Quantitation of Carbohydrate Intermediates in Glycation Systems. J. Agric. Food Chem. 2015, 63, 5911–5919. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.; Vikhnina, M.; Grishina, T.; Frolov, A. Probing Protein Glycation by Chromatography and Mass Spectrometry: Analysis of Glycation Adducts. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharm. 2014, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Smuda, M.; Henning, C.; Raghavan, C.T.; Johar, K.; Vasavada, A.R.; Nagaraj, R.H.; Glomb, M.A. Comprehensive analysis of maillard protein modifications in human lenses: Effect of age and cataract. Biochemistry 2015, 54, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Henle, T. Baking, ageing, diabetes: A short history of the Maillard reaction. Angew. Chem. Int. Ed. Engl. 2014, 53, 10316–10329. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Sun, W.; Gao, X.; Sell, D.R.; Cleary, P.A.; Lachin, J.M.; Genuth, S. Skin collagen advanced glycation endproducts (AGEs) and the long-term progression of sub-clinical cardiovascular disease in type 1 diabetes. Cardiovasc. Diabetol. 2015, 14, 118. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef]
- Skrha, J. Pathogenesis of angiopathy in diabetes. Acta Diabetol. 2003, 40 (Suppl. 2), S324–S329. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Fundamental role of pan-inflammation and oxidative-nitrosative pathways in neuropathogenesis of Alzheimer’s disease in focal cerebral ischemic rats. Am. J. Neurodegener. Dis. 2016, 5, 102–130. [Google Scholar] [PubMed]
- Chen, H.; O’Reilly, E.J.; Schwarzschild, M.A.; Ascherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Yagihashi, S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann. N. Y. Acad. Sci. 2005, 1043, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, U.; Rabbani, N.; Mullineaux, P.M.; Thornalley, P.J. Quantitative measurement of specific biomarkers for protein oxidation, nitration and glycation in Arabidopsis leaves. Plant. J. 2009, 59, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Bilova, T.; Lukasheva, E.; Brauch, D.; Greifenhagen, U.; Paudel, G.; Tarakhovskaya, E.; Frolova, N.; Mittasch, J.; Balcke, G.U.; Tissier, A.; et al. A Snapshot of the plant glycated proteome: Structural, functional, and mechanistic aspects. J. Biol. Chem. 2016, 291, 7621–7636. [Google Scholar] [CrossRef] [PubMed]
- Paudel, G.; Bilova, T.; Schmidt, R.; Greifenhagen, U.; Berger, R.; Tarakhovskaya, E.; Stöckhardt, S.; Balcke, G.U.; Humbeck, K.; Brandt, W.; et al. Changes in Arabidopsis thaliana advanced glycated proteome induced by the polyethylene glycol-related osmotic stress. J. Exp. Bot. 2016, 67, 6283–6295. [Google Scholar] [CrossRef] [PubMed]
- Bilova, T.; Paudel, G.; Shilyaev, N.; Schmidt, R.; Brauch, D.; Tarakhovskaya, E.; Milrud, S.; Smolikova, G.; Tissier, A.; Vogt, T.; et al. Global proteomic analysis of advanced glycation end products in the arabidopsis proteome provides evidence for age-related glycation hotspots. J. Biol. Chem. 2017, 292, 15758–15776. [Google Scholar] [CrossRef]
- Matamoros, M.A.; Kim, A.; Penuelas, M.; Ihling, C.; Griesser, E.; Hoffmann, R.; Fedorova, M.; Frolov, A.; Becana, M. Protein Carbonylation and Glycation in Legume Nodules. Plant Physiol. 2018, 177, 1510–1528. [Google Scholar] [CrossRef] [Green Version]
- Castellión, M.; Matiacevich, S.; Buera, P.; Maldonado, S. Protein deterioration and longevity of quinoa seeds during long-term storage. Food Chem. 2010, 121, 952–958. [Google Scholar] [CrossRef]
- Henle, T.; Walter, A.W.; Haessner, R.; Klostermeyer, H. Detection and identification of a protein-bound imidazolone resulting from the reaction of arginine residues and methylglyoxal. Z. Lebensm. Unters. Forsch. 1994, 199, 55–58. [Google Scholar] [CrossRef]
- Ahmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.; Thornalley, P.J. Assay of advanced glycation endproducts (AGEs): Surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002, 364, 1–14. [Google Scholar] [PubMed]
- Klopfer, A.; Spanneberg, R.; Glomb, M.A. Formation of arginine modifications in a model system of Nα-tert-butoxycarbonyl (Boc)-arginine with methylglyoxal. J. Agric. Food Chem. 2011, 59, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Schmidt, R.; Spiller, S.; Greifenhagen, U.; Hoffmann, R. Arginine-derived advanced glycation end products generated in peptide-glucose mixtures during boiling. J. Agric. Food Chem. 2014, 62, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Bohme, D.; Singer, D.; Frolov, A. Specific tandem mass spectrometric detection of AGE-modified arginine residues in peptides. J. Mass Spectrom. 2015, 50, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Shipanova, I.N.; Glomb, M.A.; Nagaraj, R.H. Protein modification by methylglyoxal: Chemical nature and synthetic mechanism of a major fluorescent adduct. Arch. Biochem. Biophys. 1997, 344, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J. Chromatographic assay of glycation adducts in human serum albumin glycated in vitro by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and intrinsic fluorescence. Biochem. J. 2002, 364, 15–24. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
- Henning, C.; Smuda, M.; Girndt, M.; Ulrich, C.; Glomb, M.A. Molecular basis of maillard amide-advanced glycation end product (AGE) formation in vivo. J. Biol. Chem. 2011, 286, 44350–44356. [Google Scholar] [CrossRef]
- Chevalier, F.; Chobert, J.M.; Dalgalarrondo, M.; Haertlé, T. Characterization of the Maillard reaction products of β-lactoglobulin glucosylated in mild conditions. J. Food Biochem. 2001, 25, 33–55. [Google Scholar] [CrossRef]
- Yamanaka, M.; Shirakawa, J.; Ohno, R.; Shinagawa, M.; Hatano, K.; Sugawa, H.; Arakawa, S.; Furusawa, C.; Nagai, M.; Nagai, R. Soft-shelled turtle eggs inhibit the formation of AGEs in the serum and skin of diabetic rats. J. Clin. Biochem. Nutr. 2016, 58, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Jost, T.; Zipprich, A.; Glomb, M.A. Analysis of Advanced Glycation Endproducts in Rat Tail Collagen and Correlation to Tendon Stiffening. J. Agric. Food Chem. 2018, 66, 3957–3965. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Hanke, T.; Frolov, A.; Langrock, T.; Hoffmann, R.; Fischer, C.; Schwarzenbolz, U.; Henle, T.; Born, R.; Worch, H. Modification of collagen in vitro with respect to formation of Nε-carboxymethyllysine. J. Biol. Macromol. 2009, 44, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Detection of oxidized and glycated proteins in clinical samples using mass spectrometry a user’s perspective. Biochim. Biophys. Acta 2014, 1840, 818–829. [Google Scholar] [CrossRef]
- Bottcher, C.; Roepenack-Lahaye, E.V.; Willscher, E.; Scheel, D.; Clemens, S. Evaluation of matrix effects in metabolite profiling based on capillary liquid chromatography electrospray ionization quadrupole time-of-flight massspectrometry. Anal. Chem. 2007, 79, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Murthy, U.M.; Liang, Y.; Kumar, P.P.; Sun, W.Q. Non-enzymatic protein modification by the Maillard reaction reduces the activities of scavenging enzymes in Vigna radiata. Physiol. Plant 2002, 115, 213–220. [Google Scholar] [CrossRef]
- Sun, W.Q.; Leopold, A.C. The Maillard Reaction and Oxidative Stress during Aging of Soybean Seeds. Physiol. Plant 1995, 94, 94–104. [Google Scholar] [CrossRef]
- Monnier, V.M.; Sell, D.R.; Strauch, C.; Sun, W.; Lachin, J.M.; Cleary, P.A.; Genuth, S. The DCCT Research Group The association between skin collagen glucosepane and past progression of microvascular and neuropathic complications in type 1 diabetes. J. Diabetes Complicat. 2013, 27, 141–149. [Google Scholar] [CrossRef]
- Marciniak-Darmochwal, K.; Kostyra, H. Influence of nonenzymatic glycosylation (glycation) of pea proteins (pisum sativum) on their susceptibility to enzymatic hydrolysis. J. Food Biochem. 2009, 33, 506–521. [Google Scholar] [CrossRef]
- Casey, R.; Domoney, C. Seed Proteins; Shawrey, P.R., Casey, R., Eds.; Springer: Dordrech, The Netherlands, 1999; pp. 171–208. ISBN 978-94-010-5904-6. [Google Scholar]
- Gehrig, P.M.; Krzyzaniak, A.; Barciszewski, J.; Biemann, K. Mass spectrometric amino acid sequencing of a mixture of seed storage proteins (napin) from Brassica napus, products of a multigene family. Proc. Natl. Acad. Sci. USA 1996, 93, 3647–3652. [Google Scholar] [CrossRef] [PubMed]
- Thermo ScientificTM PierceTM Carboxypeptidase Y and Resin. Available online: https://www.fishersci.co.uk/shop/products/pierce-carboxypeptidase-y-resin/11816744 (accessed on 3 December 2018).
- Proteinase from Aspergillus Melleus. Available online: https://www.sigmaaldrich.com/catalog/product/sigma/p4032?lang=en®ion=RU (accessed on 3 December 2018).
- Protease from Streptomyces Griseus. Available online: https://www.sigmaaldrich.com/catalog/product/sigma/p5147?lang=en®ion=RU (accessed on 3 December 2018).
- Breddam, K.; Ottesen, M. Determination of C-terminal sequences by digestion with serine carboxypeptidases: The influence of enzyme specificity. Curlsberg Res. Commun. 1987, 62, 55–63. [Google Scholar] [CrossRef]
- Lewis, W.S.; Schuster, S.M. Carboxypeptidase Y stability. J. Biol. Chem. 1991, 266, 20818–20822. [Google Scholar] [PubMed]
- Hashimoto, C.; Iwaihara, Y.; Chen, S.J.; Tanaka, M.; Watanabe, T.; Matsui, T. Highly-sensitive detection of free advanced glycation end-products by liquid chromatography-electrospray ionization-tandem mass spectrometry with 2,4,6-trinitrobenzene sulfonate derivatization. Anal. Chem. 2013, 85, 4289–4295. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography–electrospray–tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Ashour, A.; Thornalley, P.J. Mass spectrometric determination of early and advanced glycation in biology. Glycoconj. J. 2016, 33, 553–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glomb, M.A.; Pfahler, C. Amides are novel protein modifications formed by physiological sugars. J. Biol. Chem. 2001, 276, 41638–41647. [Google Scholar] [CrossRef] [PubMed]
- Statland, B.E.; Winkel, P.; Bokelund, H. Variation of serum iron concentration in young healthy men: Within-day and day-to-day changes. Clin. Biochem. 1976, 9, 26–29. [Google Scholar] [CrossRef]
- Greifenhagen, U.; Nguyen, V.D.; Moschner, J.; Giannis, A.; Frolov, A.; Hoffmann, R. Sensitive and site-specific identification of carboxymethylated and carboxyethylated peptides in tryptic digests of proteins and human plasma. J. Proteome Res. 2015, 14, 768–777. [Google Scholar] [CrossRef]
- Frolov, A.; Bilova, T.; Paudel, G.; Berger, R.; Balcke, G.U.; Birkemeyer, C.; Wessjohann, L.A. Early responses of mature Arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Bluher, M.; Hoffmann, R. Site-specific analysis of advanced glycation end products in plasma proteins of type 2 diabetes mellitus patients. Anal. Bioanal. Chem. 2016, 408, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | m/z | tR | LOD (fmol) | LOQ (pmol) | LDR | Slope | Intercept | R2 |
|---|---|---|---|---|---|---|---|---|
| MG-H1-d3 a | 512.23 | 12.4 | 2.5 | 0.025 | 4.0 × 104 | 5.0 × 106 | 1.0 × 107 | 0.996 |
| Parameter | Intraday Precision (n = 4) b | Inter-Day Precision (n = 4/Day) c |
|---|---|---|
| tR (min) ± SD (RSD%) | 12.3 ± 0.024 (0.196) | 12.3 ± 0.016 (0.129) |
| content (nmol/mg protein) AV ± SD (RSD%) | 87.88 ± 0.88 (1.01) | 87.35 ± 0.8 (0.92) |
| Parameter | Intraday Precision (n = 5) b | Inter-Day Precision (n = 5/Day) c |
|---|---|---|
| tR (min) ± SD (RSD%) | 12.3 ± 0.022 (0.179) | 12.3 ± 0.014 (0.117) |
| content (nmol/mg protein) AV ± SD (RSD%) | 86.72 ± 1.03 (1.185) | 86.64 ± 0.95 (1.093) |
| Parameter | Intraday Precision (n = 3) | Inter-Day Precision (n = 3/Day) b |
|---|---|---|
| tR (min) ± SD (RSD%) | 12.4 ± 0.033 (0.263) | 12.4 ± 0.0185 (0.15) |
| content (nmol mg−1protein) AV ± SD (RSD%) | 0.73 ± 0.052 (7.66) | 0.708 ± 0.037 (5.22) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonova, K.; Vikhnina, M.; Soboleva, A.; Mehmood, T.; Heymich, M.-L.; Leonova, T.; Bankin, M.; Lukasheva, E.; Gensberger-Reigl, S.; Medvedev, S.; et al. Analysis of Chemically Labile Glycation Adducts in Seed Proteins: Case Study of Methylglyoxal-Derived Hydroimidazolone 1 (MG-H1). Int. J. Mol. Sci. 2019, 20, 3659. https://doi.org/10.3390/ijms20153659
Antonova K, Vikhnina M, Soboleva A, Mehmood T, Heymich M-L, Leonova T, Bankin M, Lukasheva E, Gensberger-Reigl S, Medvedev S, et al. Analysis of Chemically Labile Glycation Adducts in Seed Proteins: Case Study of Methylglyoxal-Derived Hydroimidazolone 1 (MG-H1). International Journal of Molecular Sciences. 2019; 20(15):3659. https://doi.org/10.3390/ijms20153659
Chicago/Turabian StyleAntonova, Kristina, Maria Vikhnina, Alena Soboleva, Tahir Mehmood, Marie-Louise Heymich, Tatiana Leonova, Mikhail Bankin, Elena Lukasheva, Sabrina Gensberger-Reigl, Sergei Medvedev, and et al. 2019. "Analysis of Chemically Labile Glycation Adducts in Seed Proteins: Case Study of Methylglyoxal-Derived Hydroimidazolone 1 (MG-H1)" International Journal of Molecular Sciences 20, no. 15: 3659. https://doi.org/10.3390/ijms20153659
APA StyleAntonova, K., Vikhnina, M., Soboleva, A., Mehmood, T., Heymich, M. -L., Leonova, T., Bankin, M., Lukasheva, E., Gensberger-Reigl, S., Medvedev, S., Smolikova, G., Pischetsrieder, M., & Frolov, A. (2019). Analysis of Chemically Labile Glycation Adducts in Seed Proteins: Case Study of Methylglyoxal-Derived Hydroimidazolone 1 (MG-H1). International Journal of Molecular Sciences, 20(15), 3659. https://doi.org/10.3390/ijms20153659