Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
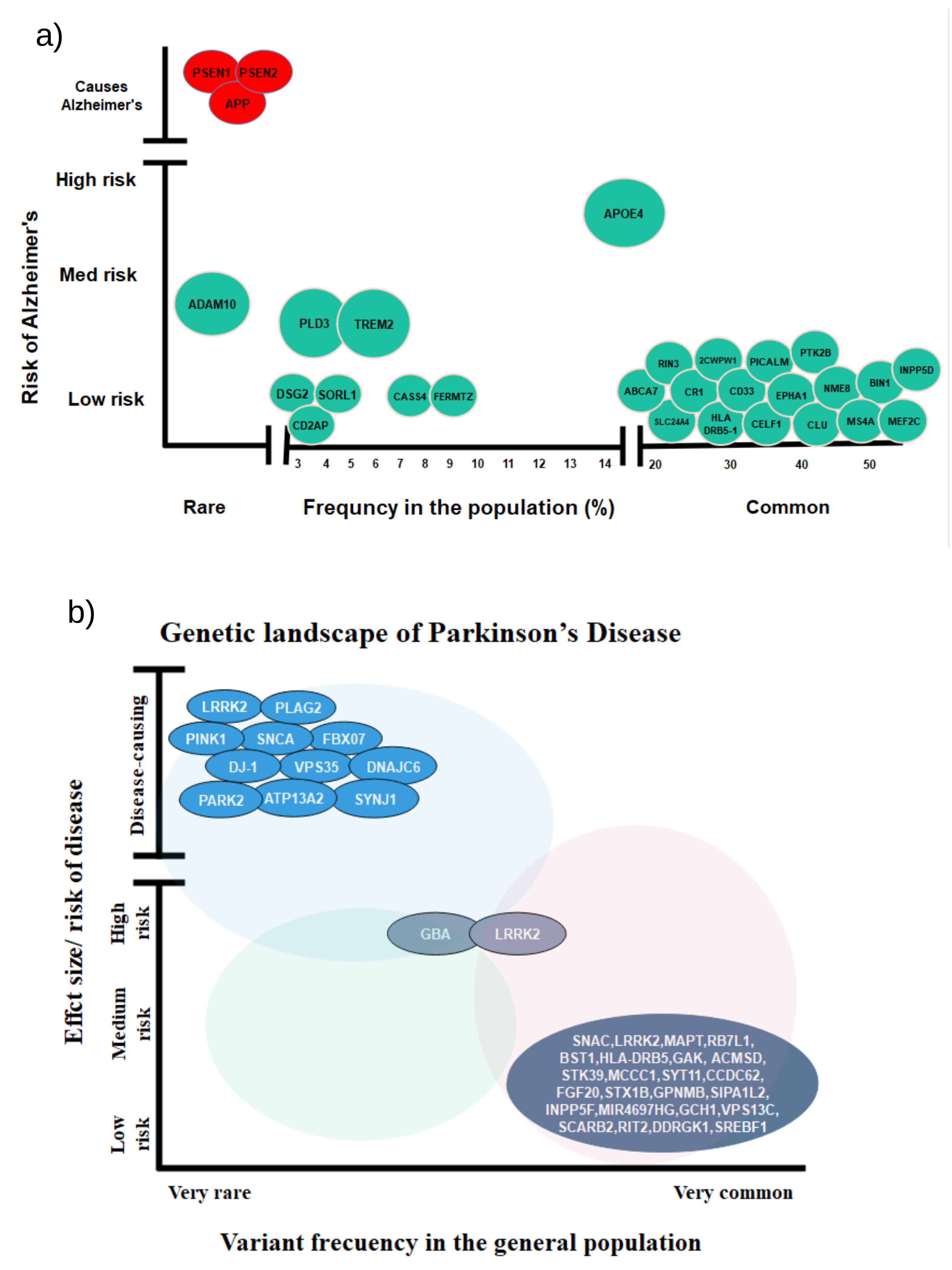
2.1. Genetic Associations of AD According to GWAS
- Lipid metabolic pathway: APOE, CLU, ABCA7
- Immune system: CLU, CR1, CD33, ABCA7, MS4A, EPHA1
- Complement system: CR1, CLU, ABCA7, CD2AP
- Endocytosis pathway: BIN1, PICLAM, CD2AP
2.2. Genetic Associations of PD According to GWAS
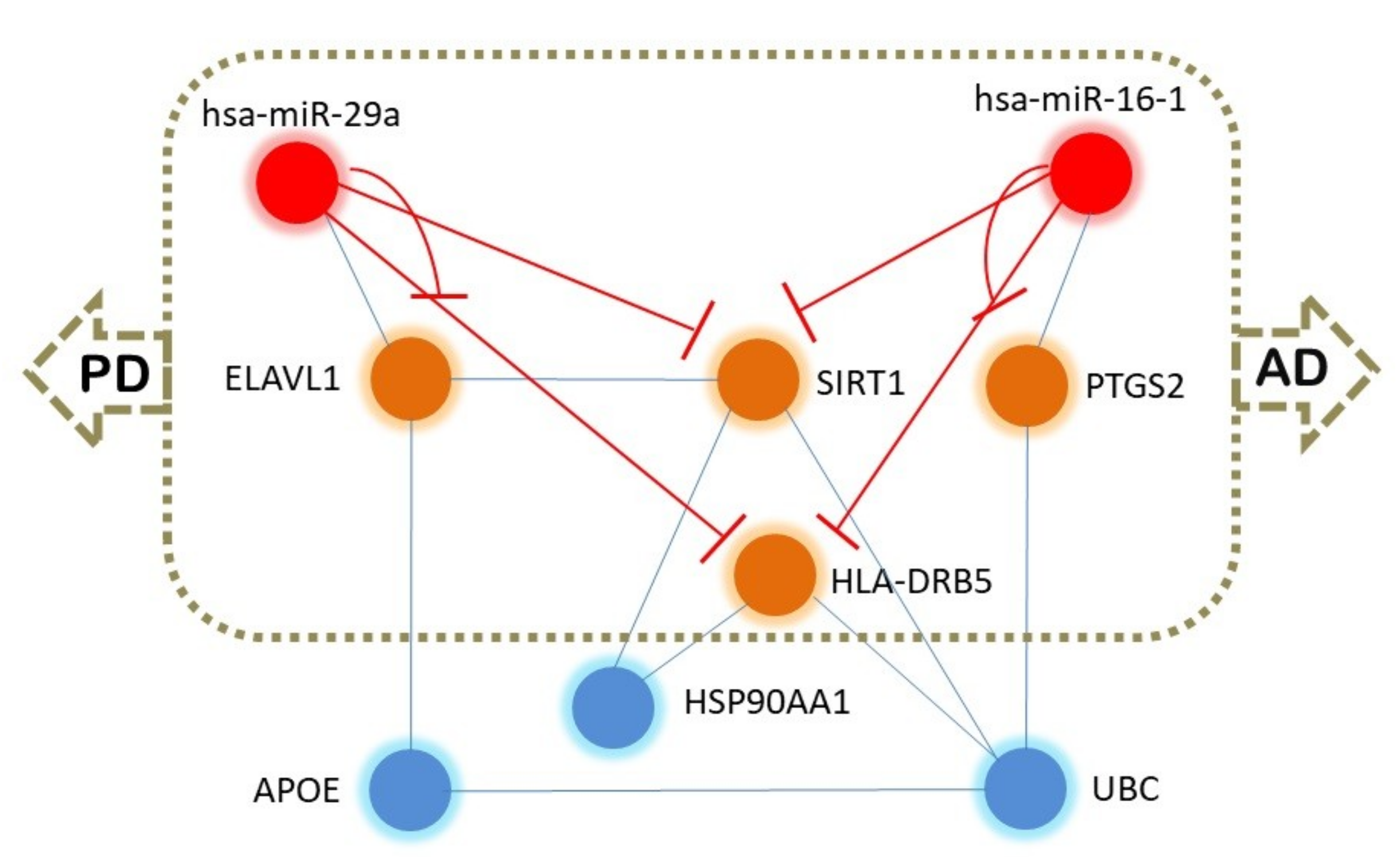
2.3. Common Regulator Genes in AD/PD
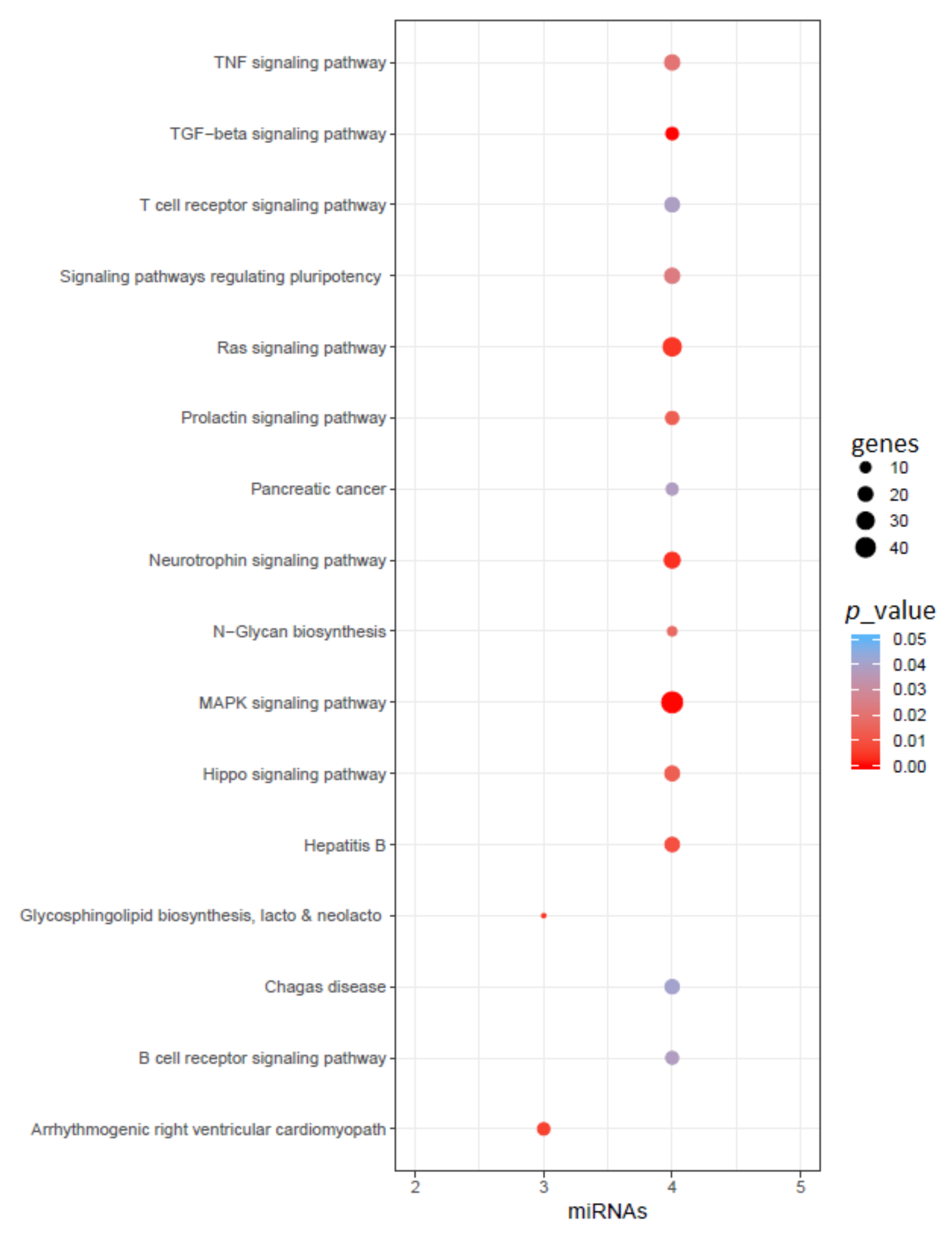
2.4. miRNAs Associated with AD and PD
2.5. Putative Epigenetic Regulation Common to AD and PD
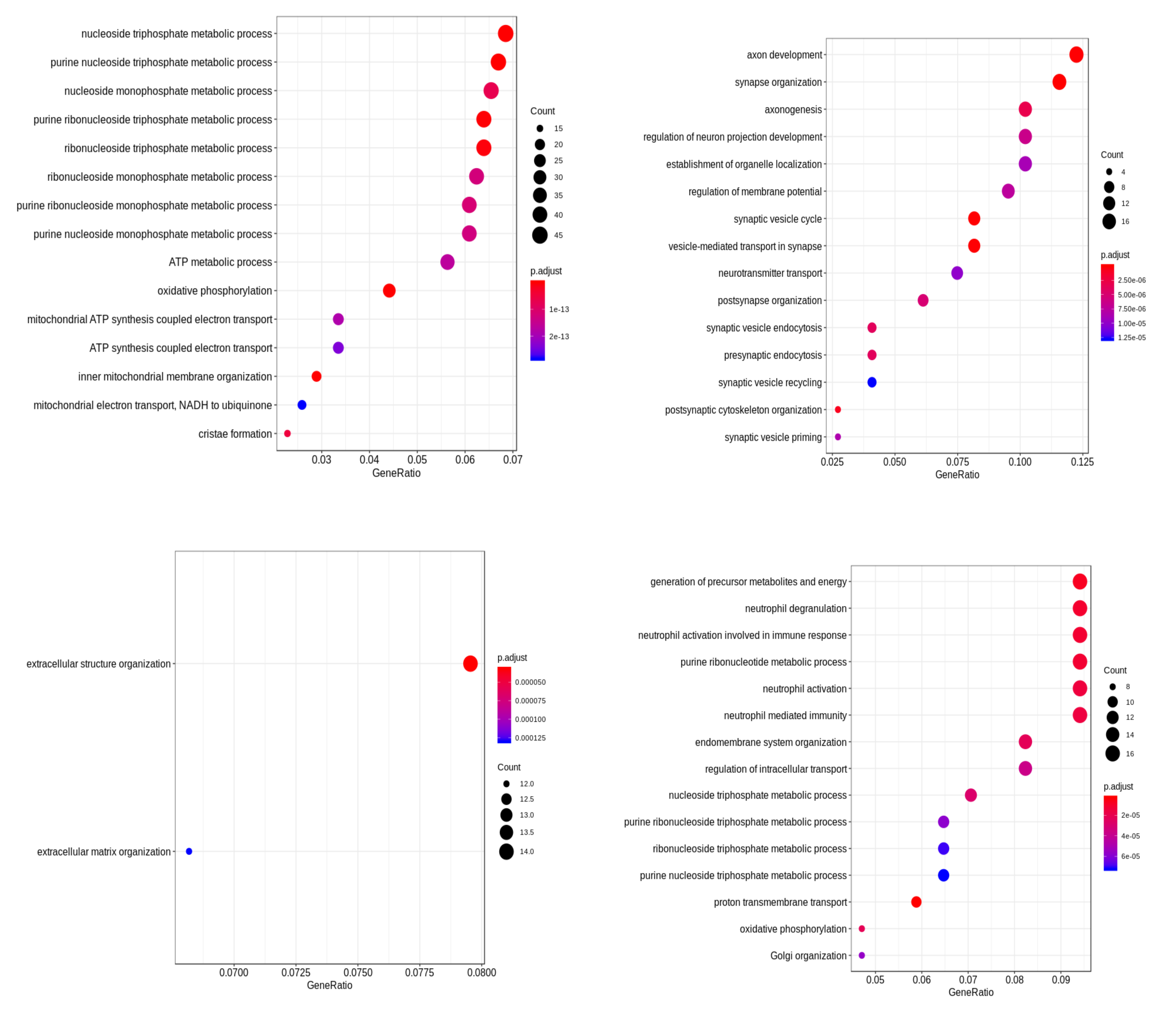
2.6. Differential Expression Analysis and Functional Enrichment on the GEO Dataset
2.7. Gene Co-Expression Network Prediction and Network Analysis on the GEO Dataset
3. Materials and Methods
3.1. Literature Mining for GWAS/miRNA Studies
3.2. Analysis on GEO Data
3.3. Gene Coexpression Network Inference Algorithm
3.4. Gene Set and Functional Similarity Analysis on the GEO Dataset
3.5. Common miRNA Identification and Pathway Analysis
4. Conclusions and Discussions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| GWAS | Genome-wide association |
| LOAD | Late-onset Alzheimer’s disease |
| miRNAs | microRNAs |
| EOAD | Early-onset Alzheimer’s disease |
| SNP | Single-nucleotide polymorphism |
| SNCA | Loci -synuclein |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPT | Microtubule-associated protein tau |
| ARVC | Arrhythmogenic right ventricular cardiomyopathy |
| DE | Differential expression |
| GEO | Gene Expression Omnibus |
| CLR | Context likelihood of relatedness |
| MRNETB | Maximum relevance minimum redundancy backward |
| GENIE3 | GEne Network Inference with Ensemble of trees |
| LIMMA | Linear Models for Microarray Data |
| DC | Distance correlation |
| GPU | Graphics processing unit |
| GRN | Gene regulatory network |
References
- Bondi, M.W.; Serody, A.B.; Chan, A.S.; Eberson-Shumate, S.C.; Delis, D.C.; Hansen, L.A.; Salmon, D.P. Cognitive and neuropathologic correlates of Stroop Color-Word Test performance in Alzheimer’s disease. Neuropsychology 2002, 16, 335. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. BioMed Res. Int. 2014, 2014, 648740. [Google Scholar] [CrossRef]
- Boller, F.; Mizutani, T.; Roessmann, U.; Gambetti, P. Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1980, 7, 329–335. [Google Scholar] [CrossRef]
- Evatt, M.L.; DeLong, M.R.; Khazai, N.; Rosen, A.; Triche, S.; Tangpricha, V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch. Neurol. 2008, 65, 1348–1352. [Google Scholar] [CrossRef]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K. The three new pathways leading to Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2011, 37, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Brás, J.; Hardy, J. SnapShot: Genetics of Alzheimer’s disease. Cell 2013, 155, 968. [Google Scholar] [CrossRef] [PubMed]
- Brás, J.; Guerreiro, R.; Hardy, J. SnapShot: Genetics of Parkinson’s disease. Cell 2015, 160, 570. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Buros, J.; Green, R.C.; Go, R.C.; Griffith, P.; Obisesan, T.O.; Shatz, R.; Borenstein, A.; et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch. Neurol. 2011, 68, 1569–1579. [Google Scholar] [CrossRef]
- Jun, G.R.; Chung, J.; Mez, J.; Barber, R.; Beecham, G.W.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Carrasquillo, M.M.; Crane, P.K.; et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimer’s Dement. 2017, 13, 727–738. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Yashin, A.I.; Fang, F.; Kovtun, M.; Wu, D.; Duan, M.; Arbeev, K.; Akushevich, I.; Kulminski, A.; Culminskaya, I.; Zhbannikov, I.; et al. Hidden heterogeneity in Alzheimer’s disease: Insights from genetic association studies and other analyses. Exp. Gerontol. 2018, 107, 148–160. [Google Scholar] [CrossRef]
- Tosto, G.; Fu, H.; Vardarajan, B.N.; Lee, J.H.; Cheng, R.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; Jimenez-Velazquez, I.Z.; Elkind, M.S.; et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Mez, J.; Chung, J.; Jun, G.; Kriegel, J.; Bourlas, A.P.; Sherva, R.; Logue, M.W.; Barnes, L.L.; Bennett, D.A.; Buxbaum, J.D.; et al. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimer’s Dement. 2017, 13, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Jun, G.; Naj, A.; Rajbhandary, R.; Vardarajan, B.N.; Wang, L.S.; Valladares, O.; Lin, C.F.; Larson, E.B.; Graff-Radford, N.R.; et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 2013, 309, 1483–1492. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Herz, J. ApoE, ApoE receptors, and the synapse in Alzheimer’s disease. Trends Endocrinol. Metab. 2017, 28, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Szumilas, M. Explaining odds ratios. J. Can. Acad. Child Adolesc. Psychiatry 2010, 19, 227. [Google Scholar]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.H.; Jossin, Y.; Liu, P.; Förster, E.; May, P.; Goffinet, A.M.; Herz, J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J. Biol. Chem. 2003, 278, 38772–38779. [Google Scholar] [CrossRef] [PubMed]
- Braskie, M.N.; Jahanshad, N.; Stein, J.L.; Barysheva, M.; McMahon, K.L.; de Zubicaray, G.I.; Martin, N.G.; Wright, M.J.; Ringman, J.M.; Toga, A.W.; et al. Common Alzheimer’s disease risk variant within the CLU gene affects white matter microstructure in young adults. J. Neurosci. 2011, 31, 6764–6770. [Google Scholar] [CrossRef] [PubMed]
- Corneveaux, J.J.; Myers, A.J.; Allen, A.N.; Pruzin, J.J.; Ramirez, M.; Engel, A.; Nalls, M.A.; Chen, K.; Lee, W.; Chewning, K.; et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 2010, 19, 3295–3301. [Google Scholar] [CrossRef]
- Erk, S.; Meyer-Lindenberg, A.; von Boberfeld, C.O.; Esslinger, C.; Schnell, K.; Kirsch, P.; Mattheisen, M.; Mühleisen, T.W.; Cichon, S.; Witt, S.H.; et al. Hippocampal function in healthy carriers of the CLU Alzheimer’s disease risk variant. J. Neurosci. 2011, 31, 18180–18184. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Yu, J.T.; Tan, L. Bridging integrator 1 (BIN1): Form, function, and Alzheimer’s disease. Trends Mol. Med. 2013, 19, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Wixler, V.; Laplantine, E.; Geerts, D.; Sonnenberg, A.; Petersohn, D.; Eckes, B.; Paulsson, M.; Aumailley, M. Identification of novel interaction partners for the conserved membrane proximal region of α-integrin cytoplasmic domains. FEBS Lett. 1999, 445, 351–355. [Google Scholar] [CrossRef]
- Chakroborty, S.; Stutzmann, G.E. Early calcium dysregulation in Alzheimer’s disease: Setting the stage for synaptic dysfunction. Sci. China Life Sci. 2011, 54, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Haroutunian, V.; DeKosky, S.T.; Sano, M.; Schadt, E.E. CR1 and the “vanishing amyloid” hypothesis of Alzheimer’s disease. Biol. Psychiatry 2013, 73, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Shulman, J.; Jagiella, J.; Cortellini, L.; Ayres, A.; Schwab, K.; Brown, D.; Silliman, S.; Selim, M.; Worrall, B.; et al. Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology 2012, 78, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crehan, H.; Holton, P.; Wray, S.; Pocock, J.; Guerreiro, R.; Hardy, J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 2012, 217, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Richard, A.C.; Rollin-Sillaire, A.; Frebourg, T.; Campion, D.; Hannequin, D. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 45–49. [Google Scholar] [CrossRef]
- Cruchaga, C.; Kauwe, J.S.; Harari, O.; Jin, S.C.; Cai, Y.; Karch, C.M.; Benitez, B.A.; Jeng, A.T.; Skorupa, T.; Carrell, D.; et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 2013, 78, 256–268. [Google Scholar] [CrossRef]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Foo, J.N.; Tan, L.C.; Irwan, I.D.; Au, W.L.; Low, H.Q.; Prakash, K.M.; Ahmad-Annuar, A.; Bei, J.; Chan, A.Y.; Chen, C.M.; et al. Genome-wide association study of Parkinson’s disease in East Asians. Hum. Mol. Genet. 2016, 26, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Consortium I.P.D.G. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Züchner, S.; Konidari, I.; Wang, G.; et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef]
- Singleton, A.; Hardy, J. The evolution of genetics: Alzheimer’s and Parkinson’s diseases. Neuron 2016, 90, 1154–1163. [Google Scholar] [CrossRef]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M.Y. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Tamouza, R.; Delord, M.; Krishnamoorthy, R.; Tzourio, C.; Mulot, C.; Nacfer, M.; Lambert, J.C.; Beaune, P.; Laurent-Puig, P.; et al. Association between Parkinson’s disease and the HLA-DRB1 locus. Mov. Disord. 2012, 27, 1104–1110. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- Donmez, G.; Guarente, L. Aging and disease: Connections to sirtuins. Aging Cell 2010, 9, 285–290. [Google Scholar] [CrossRef]
- Donmez, G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef]
- Martins, I.J. Single gene inactivation with implications to diabetes and multiple organ dysfunction syndrome. J. Clin. Epigenet. 2017, 3, 24. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in Neuroprotection by Phytochemicals: Bioactive Polyphenols Modulate Mitochondrial Apoptosis System, Function and Structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar] [CrossRef] [PubMed]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Miles, R.; Nair, A.A.; Crook, J.E.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; et al. Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: An association study with mechanistic implications. Mol. Neurodegener. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Grupe, A.; Rowland, C.; Holmans, P.; Segurado, R.; Abraham, R.; Jones, L.; Catanese, J.; Ross, D.; Mayo, K.; et al. Evidence that common variation in NEDD9 is associated with susceptibility to late-onset Alzheimer’s and Parkinson’s disease. Hum. Mol. Genet. 2007, 17, 759–767. [Google Scholar] [CrossRef]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Hu, Y.B.; Li, C.B.; Song, N.; Zou, Y.; Chen, S.D.; Ren, R.J.; Wang, G. Diagnostic value of microRNA for Alzheimer’s disease: A systematic review and meta-analysis. Front. Aging Neurosci. 2016, 8, 13. [Google Scholar] [CrossRef]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Liu, Q.Y.; Tan, M.S.; Zhang, W.; Hu, N.; Wang, Y.L.; Sun, L.; Jiang, T.; Tan, L. Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 2014, 336, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Ke, Y.D.; Humphreys, D.; Staufenbiel, M.; Ittner, L.M.; Preiss, T.; Götz, J. Neuronal microRNA deregulation in response to Alzheimer’s disease amyloid-β. PLoS ONE 2010, 5, e11070. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J.i. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Fan, G.; Zhang, J.; Wu, C.; Du, Y.; Ye, H.; Li, Z.; Wang, L.; Zhang, Z.; Zhang, L.; et al. A 9-microRNA signature in serum serves as a noninvasive biomarker in early diagnosis of Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 60, 1365–1377. [Google Scholar] [CrossRef]
- Yılmaz, Ş.G.; Erdal, M.E.; Özge, A.A.; Sungur, M.A. Can Peripheral MicroRNA Expression Data Serve as Epigenomic (Upstream) Biomarkers of Alzheimer’s Disease? OMICS J. Integr. Biol. 2016, 20, 456–461. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Martins, M.; Rosa, A.; Guedes, L.C.; Fonseca, B.V.; Gotovac, K.; Violante, S.; Mestre, T.; Coelho, M.; Rosa, M.M.; Martin, E.R.; et al. Convergence of miRNA expression profiling, α-synuclein interacton and GWAS in Parkinson’s disease. PLoS ONE 2011, 6, e25443. [Google Scholar] [CrossRef]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA profiles in Parkinson’s disease prefrontal cortex. Front. Aging Neurosci. 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Li, Y.; Wang, C.; Xu, F.; Wang, M.; Liu, Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem. Funct. 2016, 34, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Vallelunga, A.; Ragusa, M.; Di Mauro, S.; Iannitti, T.; Pilleri, M.; Biundo, R.; Weis, L.; Di Pietro, C.; De Iuliis, A.; Nicoletti, A.; et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson’s disease and Multiple System Atrophy. Front. Cell. Neurosci. 2014, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Botta-Orfila, T.; Morató, X.; Compta, Y.; Lozano, J.J.; Falgàs, N.; Valldeoriola, F.; Pont-Sunyer, C.; Vilas, D.; Mengual, L.; Fernández, M.; et al. Identification of blood serum micro-RNAs associated with idiopathic and LRRK2 Parkinson’s disease. J. Neurosci. Res. 2014, 92, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.C.T.; Barreto-Sanz, M.A.; Correia, B.R.S.; Bell, R.; Widnall, C.; Perez, L.T.; Berteau, C.; Schulte, C.; Scheller, D.; Berg, D.; et al. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget 2018, 9, 17455. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef]
- Hu, Z.; Chang, Y.C.; Wang, Y.; Huang, C.L.; Liu, Y.; Tian, F.; Granger, B.; DeLisi, C. VisANT 4.0: Integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res. 2013, 41, W225–W231. [Google Scholar] [CrossRef]
- Ma, S.L.; Tang, N.L.S.; Zhang, Y.P.; Ji, L.d.; Tam, C.W.C.; Lui, V.W.C.; Chiu, H.F.K.; Lam, L.C.W. Association of prostaglandin-endoperoxide synthase 2 (PTGS2) polymorphisms and Alzheimer’s disease in Chinese. Neurobiol. Aging 2008, 29, 856–860. [Google Scholar] [CrossRef]
- Yokota, O.; Terada, S.; Ishizu, H.; Ishihara, T.; Ujike, H.; Nakashima, H.; Nakashima, Y.; Kugo, A.; Checler, F.; Kuroda, S. Cyclooxygenase-2 in the hippocampus is up-regulated in Alzheimer’s disease but not in variant Alzheimer’s disease with cotton wool plaques in humans. Neurosci. Lett. 2003, 343, 175–179. [Google Scholar] [CrossRef]
- Teismann, P. COX-2 in the neurodegenerative process of Parkinson’s disease. Biofactors 2012, 38, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wu, Y.; Li, Y. Genetic association of cyclooxygenase-2 gene polymorphisms with Parkinson’s disease susceptibility in Chinese Han population. Int. J. Clin. Exp. Pathol. 2015, 8, 13495. [Google Scholar] [PubMed]
- Håkansson, A.; Bergman, O.; Chrapkowska, C.; Westberg, L.; Belin, A.C.; Sydow, O.; Johnels, B.; Olson, L.; Holmberg, B.; Nissbrandt, H. Cyclooxygenase-2 polymorphisms in Parkinson’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Nakatsuji, Y.; Kumanogoh, A.; Moriya, M.; Ichinose, H.; Sumi, H.; Fujimura, H.; Kikutani, H.; Sakoda, S. Loss of dopaminergic neurons by the induction of inducible nitric oxide synthase and cyclooxygenase-2 via CD40: Relevance to Parkinson’s disease. J. Neurosci. Res. 2005, 81, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Pascale, A.; Wang, J.; Ho, L.; Quattrone, A.; Gandy, S.; Haroutunian, V.; Racchi, M.; Pasinetti, G.M. nELAV proteins alteration in Alzheimer’s disease brain: A novel putative target for amyloid-β reverberating on AβPP processing. J. Alzheimer’s Dis. 2009, 16, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Soler-López, M.; Zanzoni, A.; Lluís, R.; Stelzl, U.; Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011, 21, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Helisalmi, S.; Vepsäläinen, S.; Hiltunen, M.; Koivisto, A.; Salminen, A.; Laakso, M.; Soininen, H. Genetic study between SIRT1, PPARD, PGC-1α genes and Alzheimer’s disease. J. Neurol. 2008, 255, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromol. Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Kumar, R.; Chaterjee, P.; Sharma, P.K.; Singh, A.K.; Gupta, A.; Gill, K.; Tripathi, M.; Dey, A.B.; Dey, S. Sirtuin1: A promising serum protein marker for early detection of Alzheimer’s disease. PLoS ONE 2013, 8, e61560. [Google Scholar] [CrossRef]
- García-Sierra, F.; Jarero-Basulto, J.J.; Kristofikova, Z.; Majer, E.; Binder, L.I.; Ripova, D. Ubiquitin is associated with early truncation of tau protein at aspartic acid421 during the maturation of neurofibrillary tangles in Alzheimer’s disease. Brain Pathol. 2012, 22, 240–250. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Kehl, T.; Stöckel, D.; Fehlmann, T.; Schneider, L.; Meese, E.; Lenhof, H.P.; Keller, A. miRPathDB: A new dictionary on microRNAs and target pathways. Nucleic Acids Res. 2016, gkw926. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, R6. [Google Scholar] [CrossRef]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Marshall, K.A.; et al. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 2008, 37, D885–D890. [Google Scholar] [CrossRef]
- Wang, M.; Roussos, P.; McKenzie, A.; Zhou, X.; Kajiwara, Y.; Brennand, K.J.; De Luca, G.C.; Crary, J.F.; Casaccia, P.; Buxbaum, J.D.; et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016, 8, 104. [Google Scholar] [CrossRef]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 137, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Chaitankar, V.; Ghosh, P.; Perkins, E.J.; Gong, P.; Zhang, C. Time lagged information theoretic approaches to the reverse engineering of gene regulatory networks. BMC Bioinform. 2010, 11, S19. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Marbach, D.; Roy, S.; Kellis, M. Information-Theoretic Inference of Gene Networks Using Backward Elimination. In Proceedings of the BIOCOMP’10—The 2010 International Conference on Bioinformatics &Computational Biology, Honolulu, HI, USA, 24–26 March 2010; pp. 700–705. [Google Scholar]
- Irrthum, A.; Wehenkel, L.; Geurts, P.; Huynh-Thu, V.A. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalluri, J.J.; Rana, P.; Barh, D.; Azevedo, V.; Dinh, T.N.; Vladimirov, V.; Ghosh, P. Determining causal miRNAs and their signaling cascade in diseases using an influence diffusion model. Sci. Rep. 2017, 7, 8133. [Google Scholar] [CrossRef] [PubMed]
- Nalluri, J.J.; Barh, D.; Azevedo, V.; Ghosh, P. miRsig: A consensus-based network inference methodology to identify pan-cancer miRNA-miRNA interaction signatures. Sci. Rep. 2017, 7, 39684. [Google Scholar] [CrossRef]
- Nalluri, J.; Rana, P.; Azevedo, V.; Barh, D.; Ghosh, P. Determining influential miRNA targets in diseases using influence diffusion model. In Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics, Atlanta, GA, USA, 9–12 September 2015; pp. 519–520. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2017, 46, D239–D245. [Google Scholar] [CrossRef]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2015, 44, D239–D247. [Google Scholar] [CrossRef]
- Menche, J.; Guney, E.; Sharma, A.; Branigan, P.J.; Loza, M.J.; Baribaud, F.; Dobrin, R.; Barabási, A.L. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst. Biol. Appl. 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
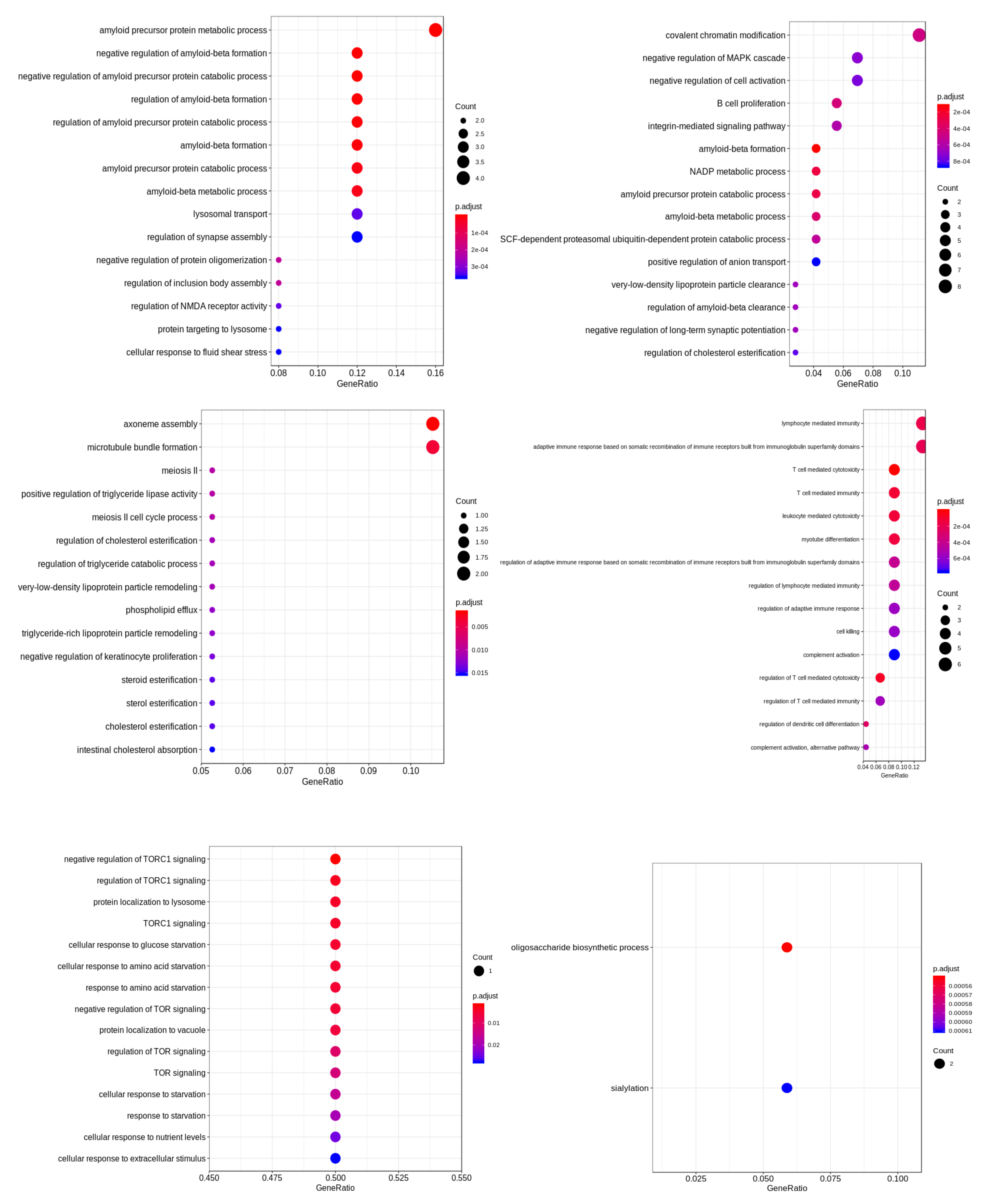{kind=link}
| Study | Ethnic Group | Sample Size | Locus | SNPs |
|---|---|---|---|---|
| [17] | African-American/ Afro-Caribbean | AD cases: 1009; Control: 6205 | CLU PICALM CR1 BIN1 CD2AP EPHA1 MS4A ABCA7 | rs2279590 rs3851179 rs6656401 rs744373 rs9349407 rs11767557 rs4938933 rs3865444 |
| [18] | European ancestry, African-American, Japanese, Israeli-Arabic | Stage 1: European ancestry: AD cases: 13,100; control: 13,220 African-American: AD cases: 1472; control: 3511 Japanese: AD cases: 951; control: 894 Israeli Arab: AD cases: 51; control: 64 Stage 2: European ancestry: AD cases: 5813; control: 20,474 | PFDN1/HBEGF USP6NL/ECHDC3 BZRAP1-AS1 NFIC | rs1116803 rs7920721 rs2632516 rs9749589 |
| [19] | European | Stage 1: AD cases: 3957; control 9682 Stage 2: AD cases: 2023; control: 2340 | TOMM40 PVRL2 APOE CLU PICALM | rs2075650 rs157580 rs6859 rs8106922 rs405509 rs11136000 rs3851179 |
| [20] | European African unspecified NR | European: 16,063 African: 2329 other: 673 | TOMM40 APOE PVRL2 APOC1 | rs2075650 rs405509 rs8106922 rs6859 rs20769449 rs12721046 rs157582 rs71352238 rs157580 rs439401 rs115881343 rs76366238 rs283815 |
| [21] | Caribbean Hispanic | AD cases: 2451; control: 2063 | TOMM40–APOE region FBXL7 CACNA2D | rs394819 rs7500204 Rs743199 |
| [22] | European | AD Cases: 71,880; control 383,378 | ADAMTS4 HESX1 CLNK CNTNAP2 ADAM10 APH1B KAT8 ALPK2 AC074212.3 | rs4575098 rs184384746 rs114360492 rs442495 rs117618017 rs59735493 rs76726049 rs76320948 |
| [23] | African-Americans | AD cases: 1825; control: 3784 | COBL SLC10A2 | rs112404845 rs16961023 |
| [24] | African-Americans | AD cases: 1968; control: 3928 | ABCA7 HMHA1 GRIN3B | rs115550680 rs115553053 rs115882880 rs145848414 |
| Study | Ethnic Group | Sample Size | Locus | SNPs |
|---|---|---|---|---|
| [25] | European | AD cases: 35,274; control: 59,163 | CR1 BIN1 INPP5D HLA-DRB1 TREM2 CD2AP NYAP1g EPHA1 PTK2B CLU SPI1h MS4A2 PICALM SORL1 FERMT2 SLC24A4 ABCA7 APOE CASS4 ECHDC3 ACE MEF2C NME8 | rs4844610 rs6733839 rs10933431 rs9271058 rs75932628 rs9473117 rs12539172 rs10808026 rs73223431 rs9331896 rs3740688 rs7933202 rs3851179 rs11218343 rs17125924 rs12881735 rs3752246 rs429358 rs6024870 rs7920721 rs138190086 rs190982 rs4723711 |
| [26] | European | Stage 1: AD cases: 17,008; control: 37,154 Stage 2: AD cases: 8572; Control: 11,312 | CR1 BIN1 CD2AP EPHA1 CLU MS4A6A PICALM ABCA7 CD33 HLA-DRB5– HLA-DRB1 PTK2B SORL1 SLC24A4- RIN3 DSG2 INPP5D MEF2C NME8 ZCWPW1 CELF1 FERMT2 | rs6656401 rs6733839 rs10948363 rs11771145 rs9331896 rs983392 rs10792832 rs4147929 rs3865444 rs9271192 rs28834970 rs11218343 rs10498633 rs8093731 rs35349669 rs190982 rs2718058 rs1476679 rs10838725 rs17125944 rs7274581 |
| Study | Ethnic Group | Sample Size | Locus | SNPs |
|---|---|---|---|---|
| [50] | European | PD cases: 5353; control: 5551 | GBA-SYT11 RAB7L1-NUCKS1 SIPA1L2 ACMSD-TMEM163 STK39 DLG2 TMEM175-GAK-DGKQ BST1 FAM47E-SCARB2 SNCA HLA-DQB1 GPNMB INPP5F DLG2 MIR4697 LRRK2 CCDC62 GCH1 TMEM229B BCKDK-STX1B MAPT RIT2 DDRGK1 FGF20 MMP16 ITGA8 | rs35749011 rs823118 rs10797576 rs6430538 rs1474055 rs12637471 rs34311866 rs11724635 rs6812193 rs356182 rs9275326 rs199347 rs117896735 rs329648 rs76904798 rs11060180 rs11158026 rs2414739 rs14235 rs17649553 rs12456492 rs8118008 rs591323 rs11868035 |
| [51] | Asian | PD cases: 5125; control: 17,604 | MCCC1 LRRK2 SNCA DLG2 | rs8180209 rs2270968 rs1384236 Rs7479949 |
| [52] | Asian | PD cases: 2011; control: 18,381 | PARK16 BST1 SNCA LRRK2 | rs16856139 rs823128 rs823122 rs947211 rs823156 rs708730 rs11240572 rs11931532 rs12645693 rs4698412 rs4538475 rs11931074 rs3857059 rs894278 rs6532194 rs1994090 rs7304279 rs4768212 rs2708453 rs2046932 |
| Study | Ethnic Group | Sample Size | Locus | SNPs |
|---|---|---|---|---|
| [53] | European | PD cases: 5333; control: 12,019 | SYT11 ACMSD STK39 MCCC1/LAMP3 GAK BST1 SNCA HLA-DRB5 LRRK2 CCDC62/HIP1R MAPT | chr1:154105678 rs6710823 rs2102808 rs11711441 chr4:911311 rs11724635 rs356219 chr6:3258820 rs1491942 rs12817488 rs2942168 |
| [54] | European | PD cases: 6476; control: 302,042 | ITPKB IL1R2 SCN3A SATB1 NCKIPSD,CDC71 ALAS1,TLR9, DNAH1,BAP1, PHF7,NISCH, STAB1ITIH3, ITIH4 ANK2, CAMK2D ELOVL7 ELOVL7 ZNF184 CTSB SORBS3, PDLIM2, C8orf58,BIN3 SH3GL2 FAM171A1 GALC COQ7 TOX3 ATP6V0A1, PSMC3IP,TUBG2 | rs4653767 rs34043159 rs353116 rs4073221 rs143918452 rs78738012 rs2694528 rs9468199 rs2740594 rs2280104 rs13294100 rs10906923 rs8005172 rs11343 rs4784227 rs601999 |
| Studies | Sample | No. of Patients | No. of Controls | Differential Expression miRNAs |
|---|---|---|---|---|
| [80] | Plasma | 31 | 37 | let-7d-5p, -7g-5p miR-15b-5p, -142-3p, -191-5p,-301a-3p,-545-3p |
| [81] | Whole Blood | 105 | 150 | miR-9, -29a, -29b, -101, -125b, -181c |
| [82] | Primary hippocampal neuron | NA | NA | miR-9, -181c, -30c, -148b, -20b let-7i |
| [83] | Brain tissues of the frontal cortex | 7 | 14 | miR-29a, -29b,-338-3p |
| [73] | Human postmortem brain specimens | NA | NA | let-7b, -7c, -7d,-7i, miR-103, -124a, -125a, -125b, -132, -134, -181a, -26a, -26b, -27a, -27b,-29a -29c, -204, -30a-5p, -7, -9 |
| [84] | Serum | 208 | 205 | novel miR-36 miR-98-5p, -885-5p, -485-5p,-483-3p,-342-3p, -3158-3p,-30e-5p, -27a-3p, -26b-3p, -191-5p, -151b, let-7g-5p,-7d-5p |
| [85] | Serum and plasma | 32 | 26 | miR-26b-3p, -125b -223, -23a |
| [74] | Brain tissue postmortem | 6 | 4 | miR-338-3p, -219-2-3p, -20a,-17, -106a, -19a, -584, -338-5p, -219-5p, -32, -34c-5p, -16, -151-5p, -181a, -181b, -485-3p, -129-5p, -143, -34a, -124, -149,-136, -138, -145, -129-3p, -381,-128, -432, -378, -29b |
| [86] | Brain tissue | 18 | 6 | miR-9, -125b, -132, -146a, -18 |
| [87] | Serum | 19 121 | 9 86 | hmiR-26a-5p, -181c-3p, 126-5p, -22-3p, 148b-5p, -106b-3p, -6119-5p, -1246, -660-5p |
| [88] | Whole blood | 172 | 109 | miR-9-5p, -106a-5p, -106b-5p, -107 |
| Studies | Sample | No. of Patients | No. of Controls | Differential expression miRNAs |
|---|---|---|---|---|
| [89] | Brain | 11 | 6 | miR-34b, miR-34c |
| [90] | Whole blood | 19 | 13 | miR-335.-374a, -199a-3p, -199b-3p, -126, -151-3p, -199a-5p, -151-5p, -29b, -147, -28-5p, -30b, -374b, -19b, -30c, -29c, -301a, -26a |
| [75] | Cerebrospinal fluid Serum | 67 | 78 | miR-132-5p, 19a-3p, -485-5p, -127-3p, -128, -409-3p, -433 -370, -431-3p, -873-3p, -121-3p, -10a, -1224-5p, -4448. miR-388-3p, -16-2-3p, -1294 -30e-3p, -30a-3p |
| [91] | Frontal cortex | 29 | 33 | miR-10b-5p |
| [92] | Serum | 138 | 112 | miR-29c,-146a, -214, and -22 |
| [93] | Whole blood | 50 | 25 | miR-24, -30c, -148b, -223, -324-3p |
| [94] | Serum | 10 65 | 10 65 | miR-29c, -19b, -92a, -16, -100 -21, 29a, -451, -19a, -181a, -484 -134, -532-5p, -223 |
| [95] | Cerebrospinal fluid | 47 | 27 | miR-1,-103a, -22, -29, -30b, -19-2,-26a, -331-5p, -153, -374 -132-5p, -119a, -485-5p, -127-3p, -151, -28, -301a, -873-3p, -136-3p -19b-3p, 10a-5p, -29c, let-7g-3p |
| [96] | Cerebrospinal fluid | 40 | 40 | miR-27a3p, -125a-5p,-151a-3p, -423-5p let-7f-5p |
| AD DE Gene | PD DE Gene (Top 50 byp-Value) |
|---|---|
| 55076, 66005, 114801, 6474, 51084 114041, 2694, 1184, 10859, 347735 53836, 3339, 254295, 51147, 147808 26050, 152573, 51412, 100289341, 27309 285194, 51678, 374920, 135228, 5788 5819, 1051, 4985, 50717, 1293, 100128927 4199, 6921, 2036, 1769, 148066, 57633 10369 | 4719, 7443, 22877, 5725, 5451 10644, 138151, 100272216, 60496, 7414 2872, 54839, 23313, 4345, 8140 404672, 55750, 10097, 81853, 5521 9201, 55209, 8905, 4190, 902 8382, 56675, 5955, 5567, 7260 5862, 11179, 30827, 400, 23242 37, 51382, 9554, 54541, 9804 801, 29887, 4839, 7994, 64175 23158, 1114, 1353, 65055, 23462 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, P.; Franco, E.F.; Rao, Y.; Syed, K.; Barh, D.; Azevedo, V.; Ramos, R.T.J.; Ghosh, P. Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2019, 20, 3730. https://doi.org/10.3390/ijms20153730
Rana P, Franco EF, Rao Y, Syed K, Barh D, Azevedo V, Ramos RTJ, Ghosh P. Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases. International Journal of Molecular Sciences. 2019; 20(15):3730. https://doi.org/10.3390/ijms20153730
Chicago/Turabian StyleRana, Pratip, Edian F. Franco, Yug Rao, Khajamoinuddin Syed, Debmalya Barh, Vasco Azevedo, Rommel T. J. Ramos, and Preetam Ghosh. 2019. "Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases" International Journal of Molecular Sciences 20, no. 15: 3730. https://doi.org/10.3390/ijms20153730
APA StyleRana, P., Franco, E. F., Rao, Y., Syed, K., Barh, D., Azevedo, V., Ramos, R. T. J., & Ghosh, P. (2019). Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases. International Journal of Molecular Sciences, 20(15), 3730. https://doi.org/10.3390/ijms20153730