Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Inbred Lines Contrasting Phenotypic and Physiological Responses to Drought Stress
2.2. Summary Inventory of Maize Filling-Kernel Proteins Identified by iTRAQ Analysis
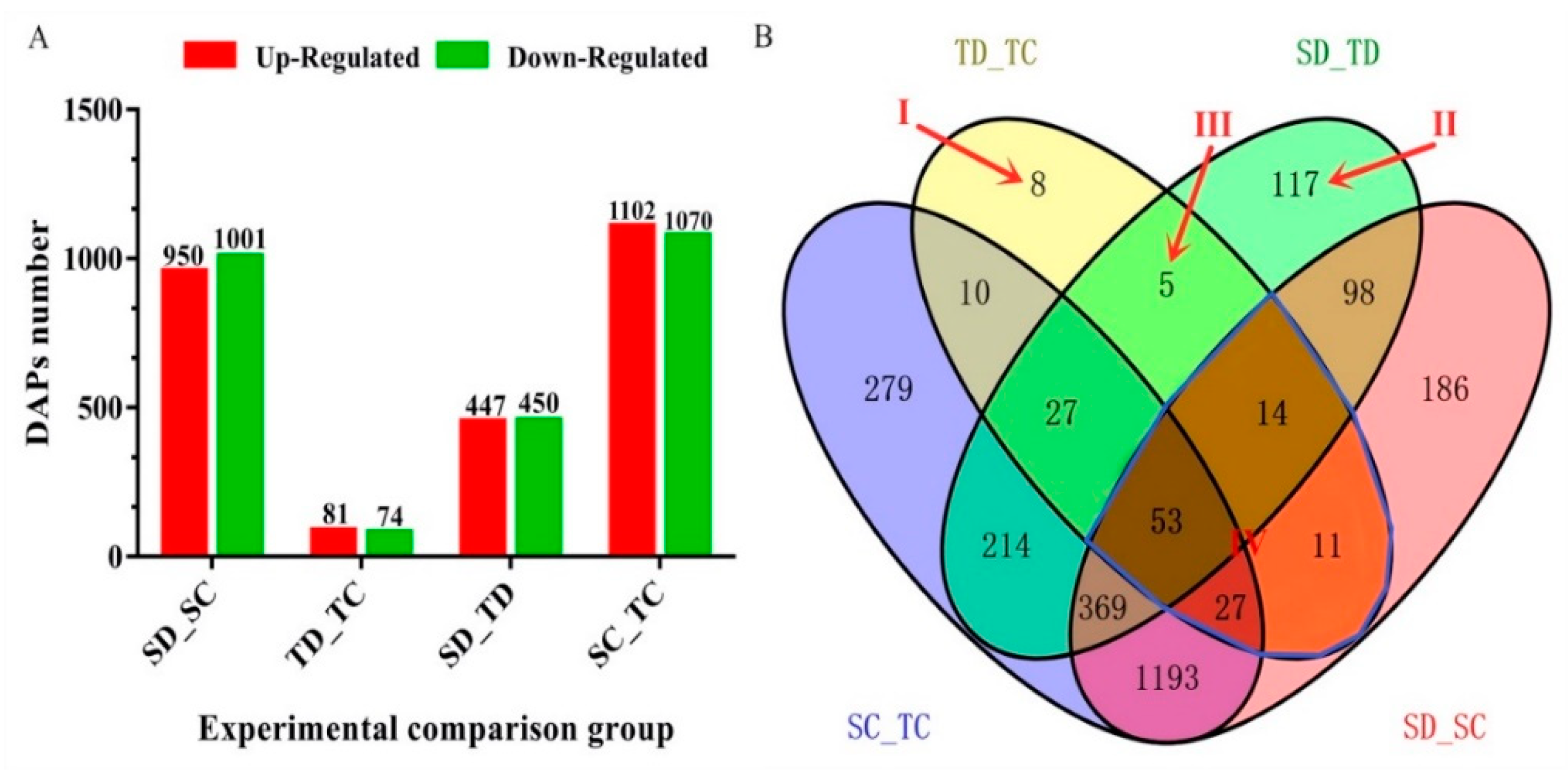
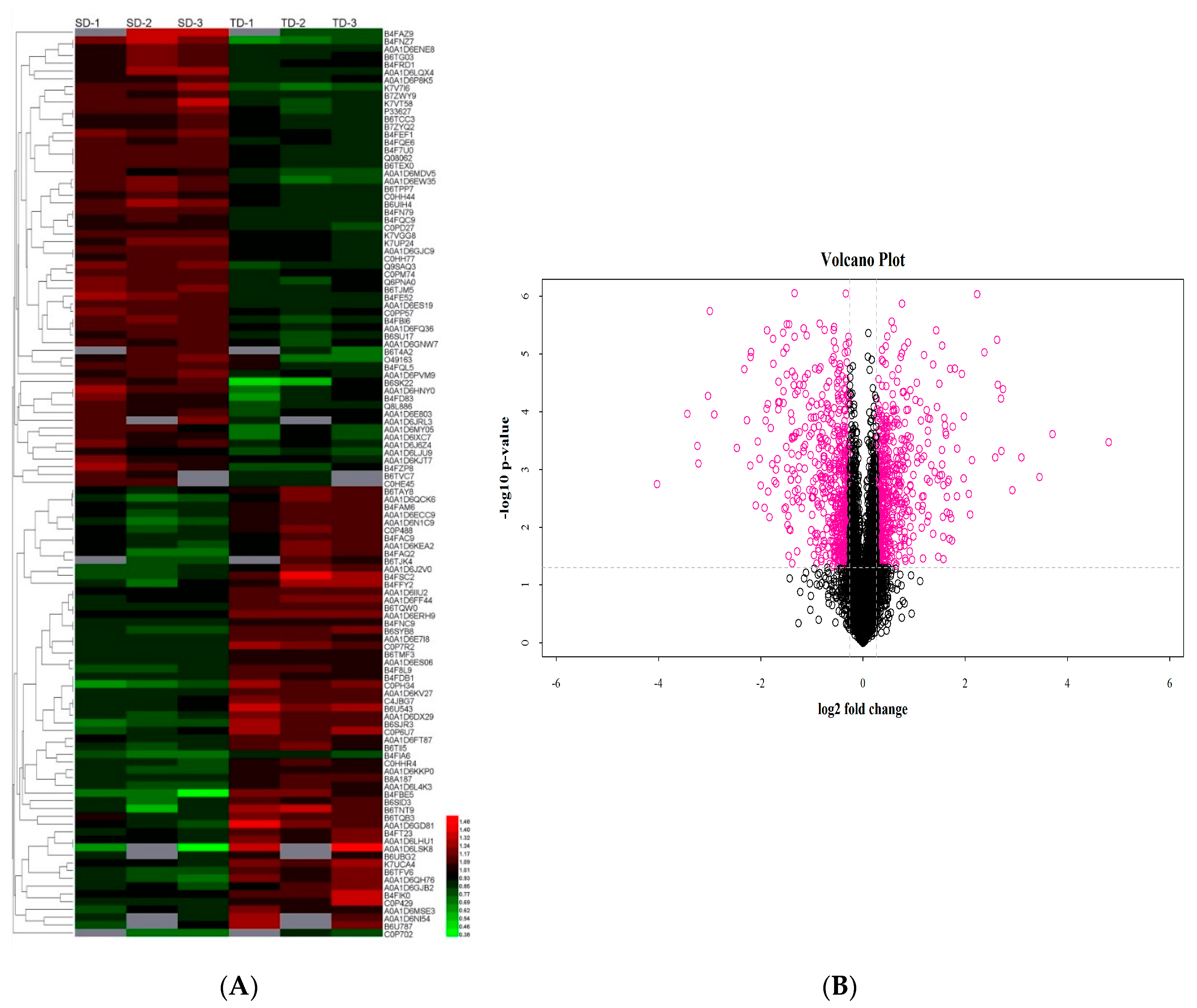
2.3. Analysis of Drought-Responsive Differentially Abundant Proteins (DAPs)
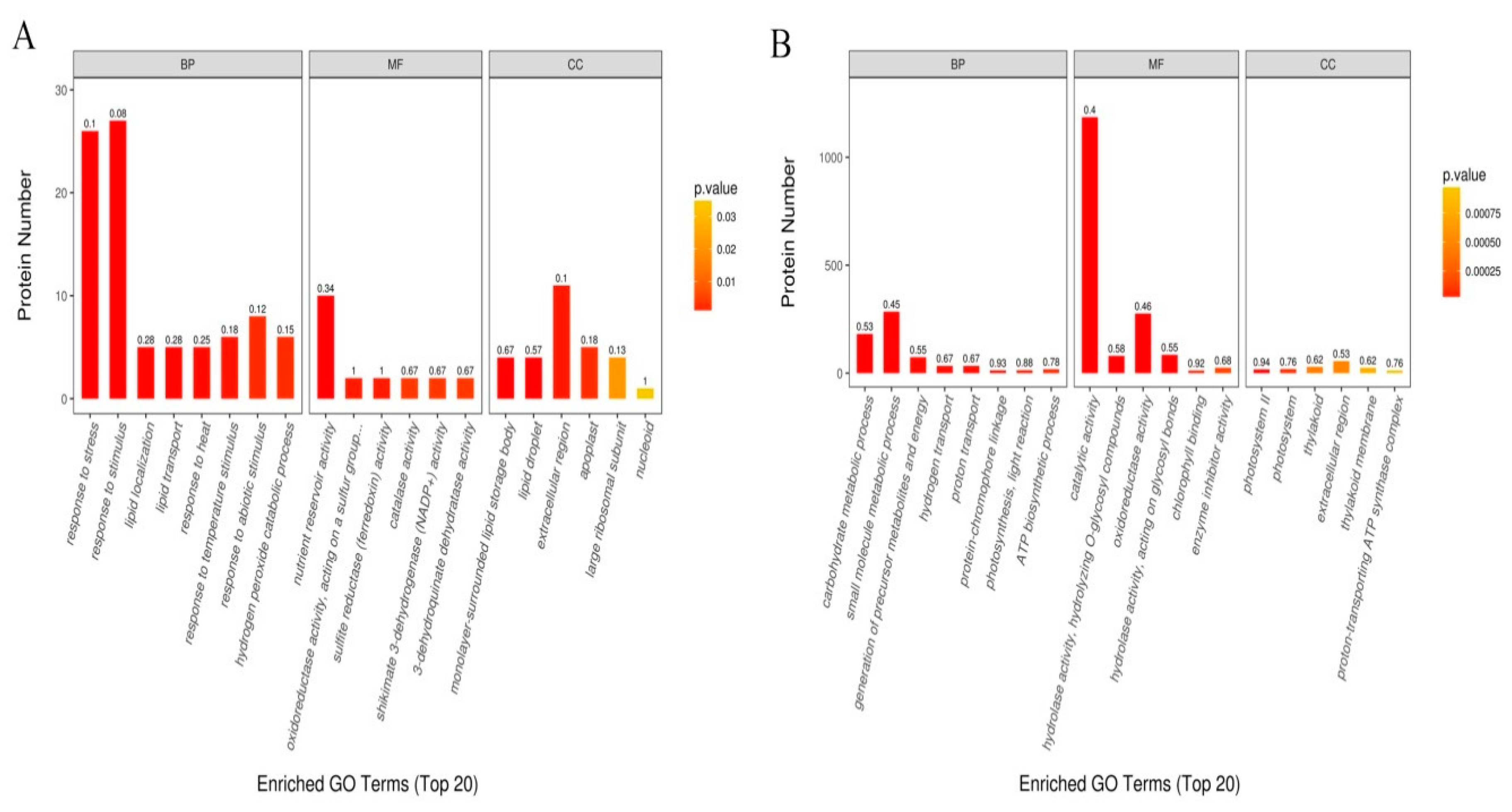
2.4. Functional Annotation and Classification of Drought-Responsive DAPs
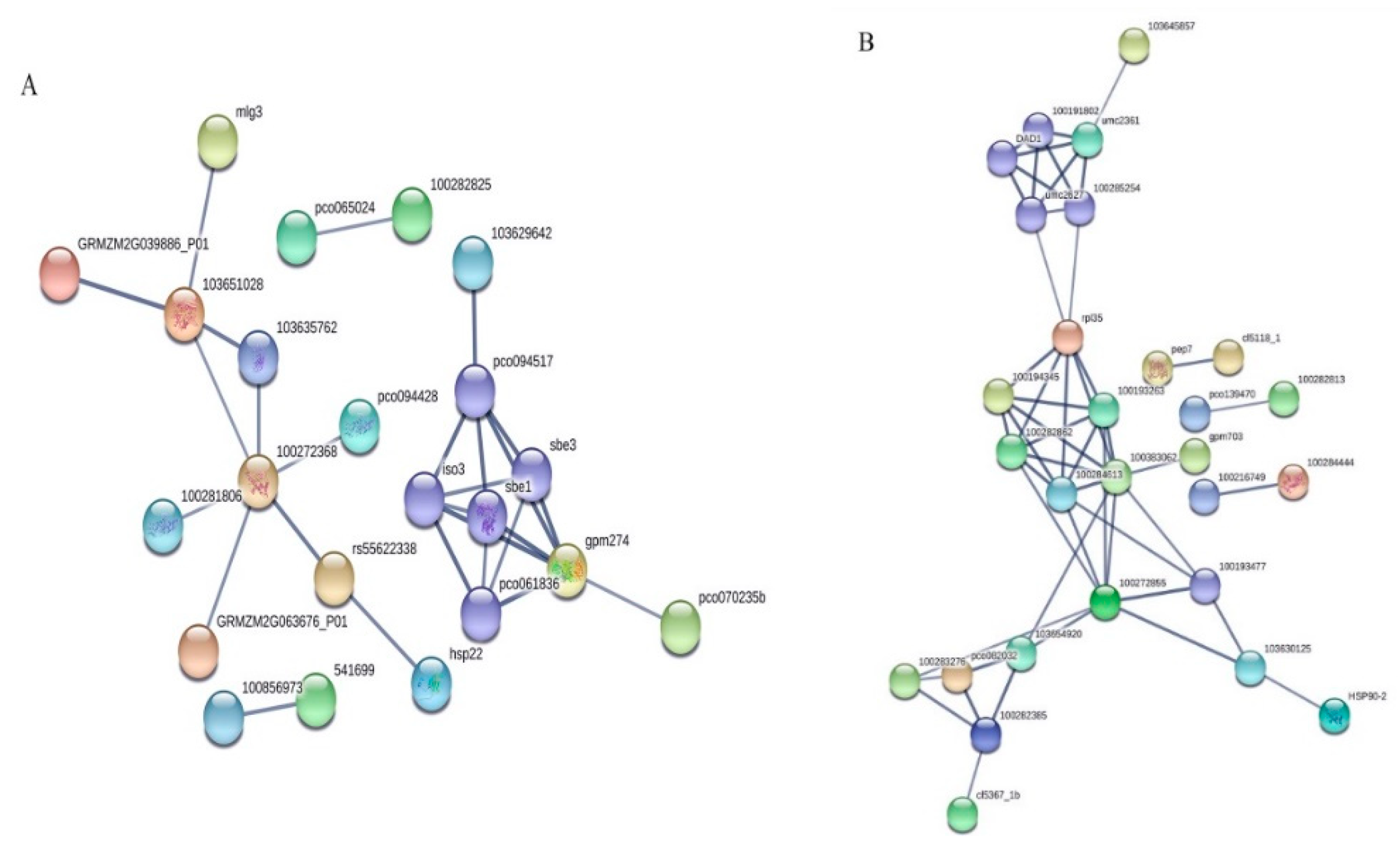
2.5. Analysis of Protein-Protein Interactions
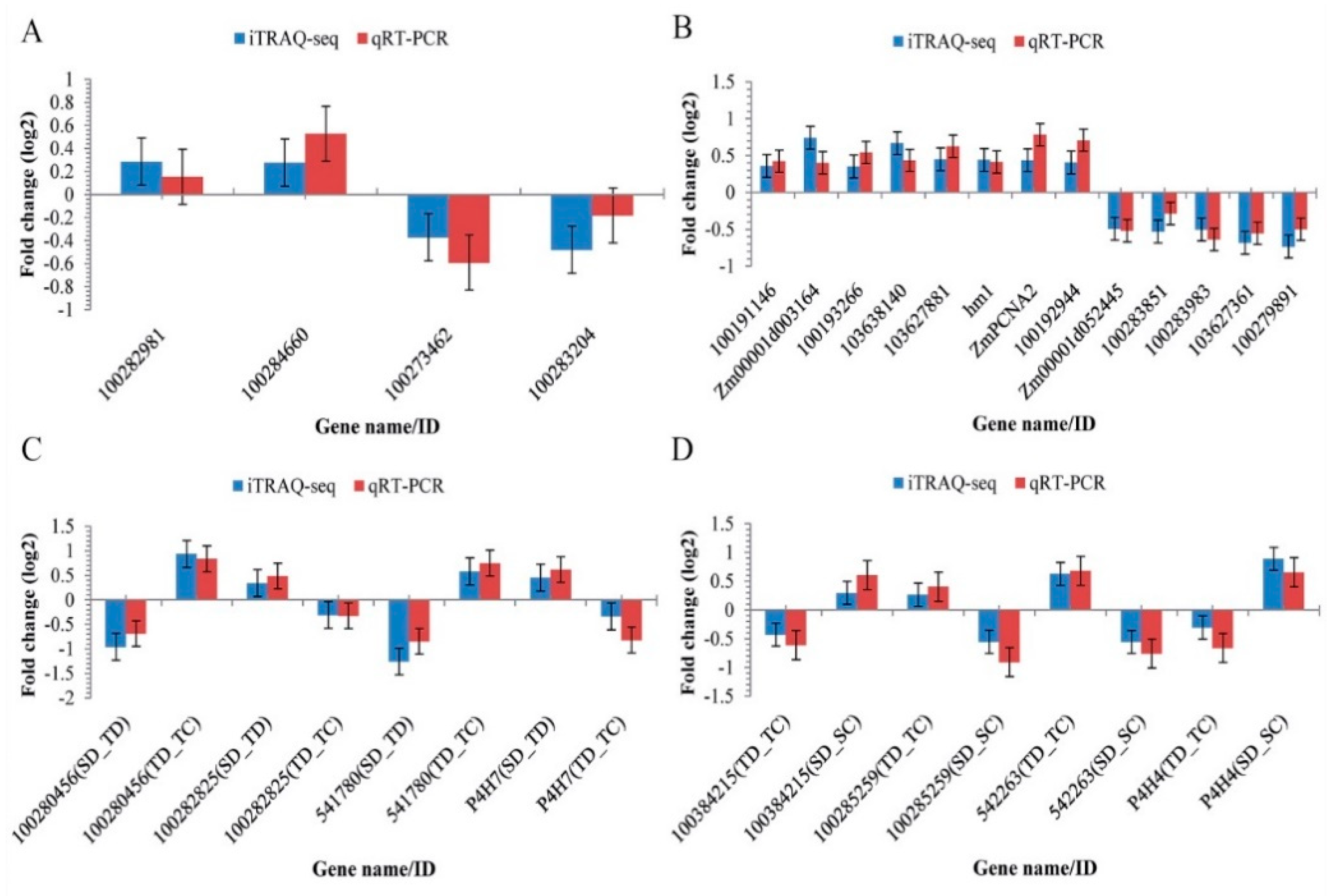
2.6. Quantitative Real-Time RT-PCR (qRT-PCR) Analysis
3. Discussion
3.1. Vivid Contrasting Phenotypic and Physiological Responses of the Two Inbred Lines to Drought Stress
3.2. Differentially-Regulated Drought-Responsive Proteins in Tolerant Line YE8112
3.2.1. Redox Post Translational Modifications (PTMs) and Epigenetic Regulation Mechanisms
3.2.2. Response to Stress- and Response to Stimuli-Related Proteins under Drought Conditions
3.2.3. Energy Metabolism and Secondary Metabolite Biosynthesis-Related Proteins under Drought
3.2.4. Seed Storage Proteins under Drought Stress
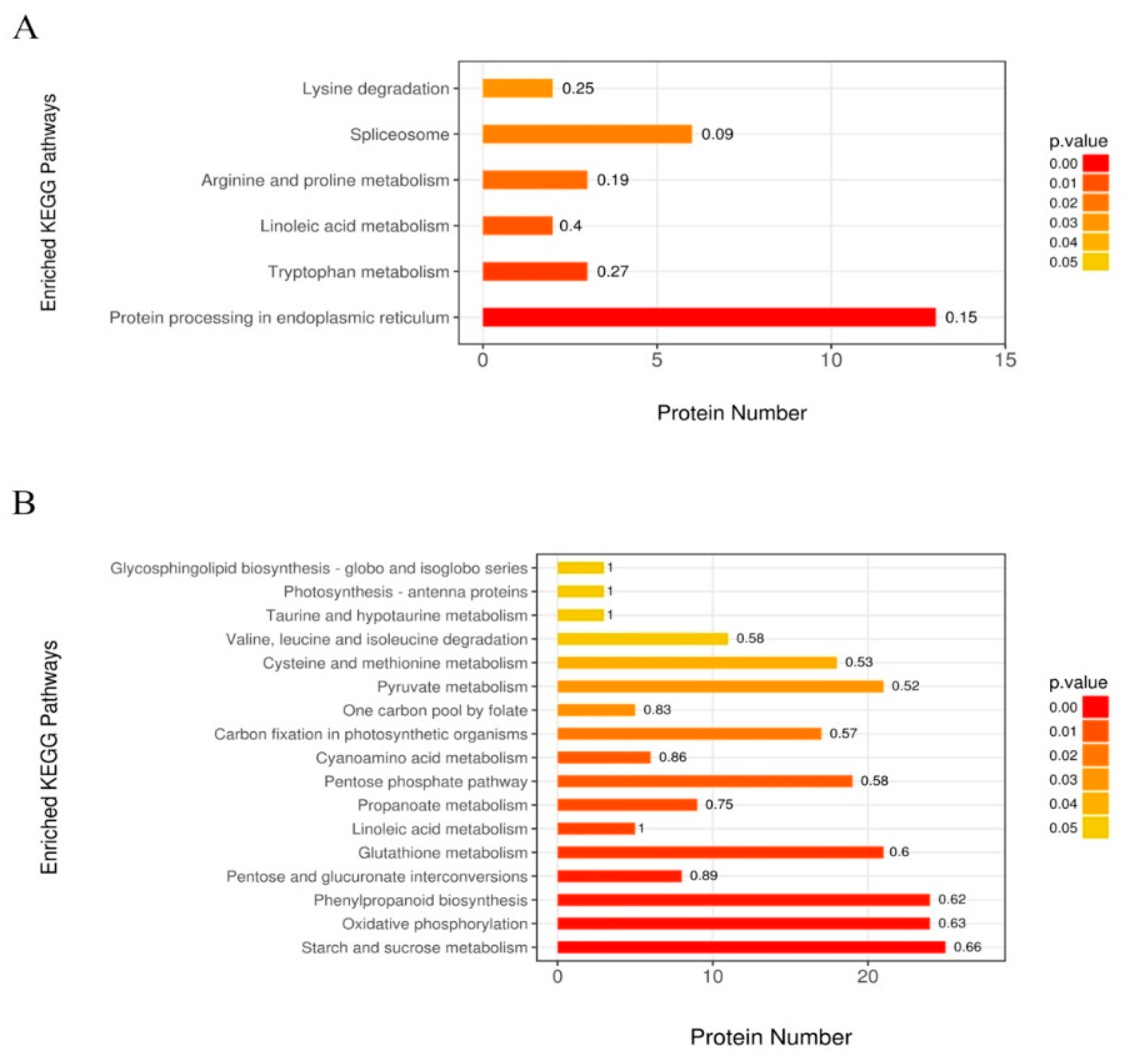
3.2.5. Most Significantly Enriched Metabolic Pathways of DAPs in Drought-Treated YE8112
3.3. Differentially-Regulated Drought-Responsive Proteins in Sensitive Line MO17
3.3.1. Plant Hormone Signal Transduction, Chaperone Activities and Protein Ubiquitination Processes under Drought Stress
3.3.2. Decreased Mitochondrial Oxidative Phosphorylation Is Vital in Reducing Cellular ROS Generation in MO17
3.3.3. Most Significantly Enriched Metabolic Pathways of DAPs in Drought-Sensitive MO17
3.4. Similarities and Differences in Drought Stress Responses between Maize Seedlings and Filling Kernels
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Phenotypic and Physiological Characterizations
4.3. Protein Extraction
4.4. Protein Digestion and Isobaric Tags for Relative and Absolute Quantification (iTRAQ) Labeling
4.5. Strong Cation Exchange (SCX) and LC-MS/MS Analysis
4.6. Protein Identification and Quantification
4.7. Functional Classification, Pathway Enrichment and Hierarchal Clustering Analyses of DAPs
4.8. RNA Extraction, cDNA Synthesis, and qRT-PCR Analysis
4.9. Physiological Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| CFI | Chalcone flavonone isomerase |
| DAP | Differentially abundant protein |
| DPP | Days post pollination |
| GAPDH/G3PDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GO | Gene ontology |
| HCTR | HC-toxin reductase |
| HDAC | Histone deacetylase |
| HSPs; sHSPs | Heat shock proteins; small HSPs |
| iTRAQ | Isobaric tags for relative and absolute quantification |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LEA | Late embryogenesis abundant (proteins) |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| MDA | Malondialdehyde |
| MDH | Malate dehydrogenate |
| NRA | Nutrient reservoir activity |
| POD | Peroxidases |
| PTMs | Post translational modifications |
| PPER | Protein processing in the endoplasmic reticulum |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| ROS | Reactive oxygen species |
| UCHs | Ubiquitin carboxyl-terminal hydrolases |
References
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, T.K.; Bashir, T.; Hashem, A.; Abd_Allah, E.F. Systems biology approach in plant abiotic stresses. Plant Physiol. Biochem. 2017, 121, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, T.; Fountain, J.C.; Scully, B.T.; Lee, R.D.; Kemerait, R.C.; Chen, S.; Guo, B. Protein profiles reveal diverse responsive signaling pathways in kernels of two maize inbred lines with contrasting drought sensitivity. Int. J. Mol. Sci. 2014, 15, 18892–18918. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Cruz de Carvalho, M.H.; Torres-Jerez, I.; Kang, Y.; Allen, S.N.; Huhman, D.V.; Tang, Y.; Murray, J.; Sumner, L.W.; Udvardi, M.K. Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after rewatering. Plant Cell Environ. 2014, 37, 2553–2576. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Nagler, M.; Roustan, V.; Lyon, D.; Bachmann, G.; Postl, W.; Schröfl, A.; Desai, N.; Varshney, R.K.; et al. Comprehensive tissue-specific proteome analysis of drought stress responses in Pennisetum glaucum (L.) R. Br. (Pearl millet). J. Proteom. 2016, 143, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Edmeades, G.O. Progress in Achieving and Delivering Drought Tolerance in Maize-An Update; ISAA: Ithaca, NY, USA, 2013; pp. 1–39. [Google Scholar]
- Feller, U.; Vaseva, I.I. Extreme climatic events: Impacts of drought and high temperature on physiological processes in agronomically important plants. Front. Environ. Sci. 2014, 2, 39. [Google Scholar] [CrossRef]
- Thirunavukkarasu, N.; Sharma, R.; Singh, N.; Shiriga, K.; Mohan, S.; Mittal, S.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; et al. Genomewide expression and functional interactions of genes under drought stress in maize. Int. J. Genet. 2017, 2017, 2568706. [Google Scholar] [CrossRef]
- Aslam, M.; Maqbool, M.A.; Cengiz, R. Drought stress in maize (Zea mays L.): Effects, resistance mechanisms, global achievements and biological strategies for improvement. In SpringerBriefs in Agriculture; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-25440-1. [Google Scholar]
- Campos, H.; Cooper, M.; Habben, J.E.; Edmeades, G.O.; Schussler, J.R. Improving drought tolerance in maize: A view from industry. Field Crops Res. 2004, 90, 19–34. [Google Scholar] [CrossRef]
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.F.; Jakson, B.S.; Kiniry, J.R.; Arkin, G.F. Water deficit timing effects on yield components in maize. Agron. J. 1989, 81, 61–65. [Google Scholar] [CrossRef]
- Chapman, S.C.; Edmeades, G.O.; Crossa, J. Pattern analysis of grains from selection for drought tolerance in tropical maize population. In Plant Adaptation and Crop Improvement; Cooper, M., Hammer, G.L., Eds.; CAB INTERNATIONAL: Walling Ford, UK, 1996; pp. 513–527. [Google Scholar]
- Doll, N.M.; Depe‘ge-Fargeix, N.; Rogowsky, P.M.; Widiez, T. Signaling in Early Maize Kernel Development. Mol. Plant 2017, 10, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, W.B.; Edmeades, G.O.; Barker, T.C. Molecular and physiological approaches to maize improvement for drought tolerance. J. Exp. Bot. 2002, 53, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fountain, J.C.; Ji, P.; Ni, X.; Chen, S.; Lee, R.D.; Kemerait, R.C.; Guo, B. Deciphering drought-induced metabolic responses and regulation in developing maize kernels. Plant Biotechnol. J. 2018, 16, 1616–1628. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, J.; Lee, D.R.; Scully, B.T.; Guo, B.Z. Monitoring the expression of maize genes in developing kernels under drought stress using oligo-microarray. J. Integr. Plant Biol. 2010, 52, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Sawant, K. Drought stress adaptation: Metabolic adjustment and regulation of gene expression. Plant Breed. 2013, 132, 21–32. [Google Scholar] [CrossRef]
- Priya, M.; Siddique, K.H.M.; Dhankhar, O.P.; Prasad, P.V.V.; Rao, B.H.; Nair, R.M.; Nayyar, H. Molecular breeding approaches involving physiological and reproductive traits for heat tolerance in food crops. Ind. J. Plant Physiol. 2018, 23, 697–720. [Google Scholar] [CrossRef]
- Osmolovskaya, N.; Shumilina, J.; Kim, A.; Didio, A.; Grishina, T.; Bilova, T.; Keltsieva, O.A.; Zhukov, V.; Tikhonovich, I.; Tarakhovskaya, E.; et al. Methodology of Drought Stress Research: Experimental Setup and Physiological Characterization. Int. J. Mol. Sci. 2018, 19, 4089. [Google Scholar] [CrossRef]
- Maiti, R.K.; Maiti, L.E.; Sonia, M.; Maiti, A.M.; Maiti, M.; Maiti, H. Genotypic variability in maize (Zea mays L.) for resistance to drought and salinity at the seedling stage. J. Plant Physiol. 1996, 148, 741–744. [Google Scholar] [CrossRef]
- Ahmadi, A.; Emam, Y.; Pessarakli, M. Biochemical changes in maize seedlings exposed to drought stress conditions at different nitrogen levels. J. Plant Nutr. 2010, 33, 541–556. [Google Scholar] [CrossRef]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H.; et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lei, L.; Lai, J.; Zhao, H.; Song, W. Effects of drought stress and water recovery on physiological responses and gene expression in maize seedlings. BMC Plant Biol. 2018, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Kosova, K.; Vitamvas, P.; Prasil, I.T.; Renaut, J. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ning, F.; Zhang, Q.; Wu, X.; Wang, W. Enhancing omics research of crop responses to drought under field conditions. Front. Plant Sci. 2017, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.T.; Lim, Y.S.; Lau, S.E. Proteomics in commercial crops: An overview. J. Protozool. 2017, 169, 176–188. [Google Scholar] [CrossRef]
- Komatsu, S. Plant Proteomic Research 2.0: Trends and Perspectives. Int. J. Mol. Sci. 2019, 20, 2495. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Smith, I.H.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1995, 13, 19–50. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Cereal crop proteomics: Systemic analysis of crop drought stress responses towards marker-assisted selection breeding. Front. Plant Sci. 2017, 8, 757. [Google Scholar] [CrossRef]
- Labuschagne, M.T. A review of cereal grain proteomics and its potential for sorghum improvement. J. Cereal Sci. 2018, 84, 151–158. [Google Scholar] [CrossRef]
- Kalisa, S.K.; Wu, H.F. Recent developments in nanoparticle-based MALDI mass spectrometric analysis of phosphoproteomes. Microchim. Acta 2014, 181, 853–864. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Sarkar, A.; Righetti, P.G.; Pedreschi, R.; Carpentier, S.; Wang, T.; Barkla, B.J.; Kohli, A.; Ndimba, B.K.; Bykova, N.V.; et al. A decade of plant proteomics and mass spectrometry: Translation of technical advancements to food security and safety issues. Mass Spectrom. Rev. 2013, 32, 335–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, D.; Wu, D.; Liu, J.; Li, D.; Xu, C.; Li, S.; Li, P.; Zhang, H.; Liu, X.; Jiang, C.; et al. Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS ONE 2015, 10, e0116697. [Google Scholar] [CrossRef] [PubMed]
- Matros, A.; Kaspar, S.; Witzel, K.; Mock, H.P. Recent progress in liquid chromatography-based separation and label-free quantitative plant proteomics. Phytochemistry 2011, 72, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wu, L.; Zhao, F.; Zhang, D.; Li, N.; Zhu, G.; Li, C.; Wang, W. Phosphoproteomic analysis of the response of maize leaves to drought, heat and their combination stress. Front. Plant Sci. 2015, 6, 298. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, W. Increasing confidence of proteomics data regarding the identification of stress-responsive proteins in crop plants. Front. Plant Sci. 2016, 7, 702. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Yang, H.; Wang, W.; Wu, J.; Hu, X. Quantitative proteomic analyses identify aba-related proteins and signal pathways in maize leaves under drought conditions. Front. Plant Sci. 2016, 7, 1827. [Google Scholar] [CrossRef]
- Li, G.K.; Gao, J.; Peng, H.; Shen, Y.O.; Ding, H.P.; Zhang, Z.M.; Pan, G.T.; Lin, H.J. Proteomic changes in maize as a response to heavy metal (lead) stress revealed by iTRAQ quantitative proteomics. Genet. Mol. Res. 2016, 15, 1–14. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, Y.; Wang, Y.; Shi, Z.; Zhang, P.; Zhang, Y.; Song, W.; Zhao, J. Comparative proteomics of contrasting maize genotypes provides insights into salt-stress tolerance mechanisms. J. Proteome Res. 2018, 17, 141–153. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef] [PubMed]
- Zenda, T.; Liu, S.T.; Wang, X.; Liu, G.; Jin, H.Y.; Dong, A.Y.; Yang, Y.T.; Duan, H.J. Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines. Int. J. Mol. Sci. 2019, 20, 1268. [Google Scholar] [CrossRef] [PubMed]
- Edmeades, G.O.; Bolaños, J.; Elings, A.; Ribaut, J.M.; Bänziger, M.; Westgate, M.E. The Role and Regulation of the Anthesis-Silking Interval in Maize. In Physiology and Modeling Kernel Set in Maize; Westgate, M.E., Boote, K.J., Eds.; CSSA Special Publication: WI, USA, 2000; Volume 29, pp. 43–73. [Google Scholar]
- Yadav, R.S.; Hash, C.T.; Bidinger, F.R.; Devos, K.M.; Howarth, C.J. Genomic regions associated with grain yield and aspects of post flowering drought tolerance in pearl millet across environments and tester background. Euphytica 2004, 136, 265–277. [Google Scholar] [CrossRef]
- Anjum, S.A.; Xie, X.Y.; Wang, L.C.; Saleem, M.F.; Man, C.; Lei, W. Morphological, physiological and biochemical responses of plants to drought stress. Afr. J. Agric. Res. 2011, 6, 2026–2032. [Google Scholar]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef]
- Mallick, N.; Mohn, F.H.; Soeder, C.J. Impact of physiological stresses on nitric oxide formation by green alga, scenedesmus obliquus. J. Microbiol. Biotechnol. 2000, 110, 300–307. [Google Scholar]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 10, 26. [Google Scholar] [CrossRef]
- Reddy, P.S.; Jogeswar, G.; Rasineni, G.K.; Maheswari, M.; Reddy, A.R.; Varshney, R.K.; Kishor, P.B.K. Proline over-accumulation alleviates salt stress and protects photosynthetic and antioxidant enzyme activities in transgenic sorghum [Sorghum bicolor (L.) Moench]. Plant Physiol. Biochem. 2015, 94, 104–113. [Google Scholar] [CrossRef]
- Kaur, G.; Asthir, B. Proline: A key player in plant abiotic stress tolerance. Biol. Plant. 2015, 59, 609–619. [Google Scholar] [CrossRef]
- Davey, M.W.; Stals, E.; Panis, B.; Keulemans, J.; Swennen, R.L. High-throughput determination of malondialdehyde in plant tissues. Anal. Biochem. 2005, 347, 201–207. [Google Scholar] [CrossRef]
- Guo, L.; Devaiah, S.P.; Narasimhan, R.; Pan, X.; Zhang, Y.; Zhang, W.; Wang, X. Cytosolic glyceraldehyde-3-phosphate dehydrogenases interact with phospholipase D delta to transduce hydrogen peroxide signals in the Arabidopsis response to stress. Plant Cell 2012, 24, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.R.; Huang, Y.W.; Liou, M.R.; Wang, R.Y.; Hu, C.C.; Tsai, C.H.; Meng, M.; Lin, N.S.; Hsu, Y.H. Glyceraldehyde 3-phosphate dehydrogenase negatively regulates the replication of Bamboo mosaic virus and its associated satellite RNA. J. Virol. 2011, 85, 8829–8840. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, M.; Fermani, S.; Costa, A.; Lemaire, S.D.; Trost, P. Plant cytoplasmic GAPDH: Redox post-translational modifications and moonlighting properties. Front. Plant Sci. 2013, 4, 450. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, X.Y.; Xin, M.M.; Du, J.K.; Hu, Z.R.; Peng, H.R.; Rossi, V.; Sun, Q.X.; Ni, Z.F.; Yao, Y.Y. Genome-Wide Mapping of Targets of Maize Histone Deacetylase HDA101 Reveals Its Function and Regulatory Mechanism during Seed Development. Plant Cell 2016, 28, 629–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frydman, J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem. 2001, 70, 603–647. [Google Scholar] [CrossRef]
- Volkov, R.A.; Panchuk, I.; Mullineaux, P.M.; Schöffl, F. Heat stress-induced H2O2 is required for effective expression of heat shock genes in Arabidopsis. Plant Mol. Biol. 2006, 61, 733–746. [Google Scholar] [CrossRef]
- Tripathy, B.C.; Oelmüller, R. Reactive oxygen species generation and signaling in plants. Plant Signal. Behav. 2016, 7, 1621–1633. [Google Scholar] [CrossRef]
- Buchner, J. Hsp90 & Co.—A holding for folding. Trends Biochem. Sci. 1999, 24, 136–141. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef]
- Bakthisaran, R.; Tangirala, R.; Rao, C.M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta 2015, 1854, 291–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.S.; Zhu, H.B.; Jin, G.L.; Liu, H.L.; Wu, W.R.; Zhu, J. Genome-scale identification and analysis of LEA genes in rice (Oryza sativa L.). Plant Sci. 2006, 172, 414–420. [Google Scholar] [CrossRef]
- Tolleter, D.; Hincha, D.K.; Macherel, D. A mitochondrial late embryogenesis abundant protein stabilizes model membranes in the dry state. Biochim. Biophys. Acta 2010, 1798, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Lee, J.-S.; Kim, J.-T.; Kwon, Y.S.; Bae, D.-W.; Bae, H.H.; Son, B.-Y.; Baek, S.-B.; Kwon, Y.-U.; Woo, M.-O.; et al. Physiological and Proteomics Analysis of the response to drought stress in an inbred Korean maize line. Plant Omics 2015, 8, 159–168. [Google Scholar]
- Hayashi, M.; Takahashi, H.; Tamura, K.; Huang, J.; Yu, L.H.; Kawai-Yamada, M.; Tezuka, T.; Uchimiya, H. Enhanced dihydroflavonol-4-reductase activity and NAD homeostasis leading to cell death tolerance in transgenic rice. Proc. Natl. Acad. Sci. USA 2005, 102, 7020–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shewry, P.R.; Napier, J.A.; Tatham, A.S. Seed Storage Proteins: Structures’ and Biosynthesis. Plant Cell 1995, 7, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Bicudo, T.C.; Forato, L.A.; Batista, L.A.R.; Colnago, L.A. Study of the conformation of γ-zeins in purified maize protein bodies by FTIR and NMR spectroscopy. Anal. Bioanal. Chem. 2005, 383, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Holding, D.R. Recent advances in the study of prolamin storage protein organization and function. Front. Plant Sci. 2014, 5, 276. [Google Scholar] [CrossRef] [Green Version]
- Radwanski, E.R.; Last, R.L. Tryptophan Biosynthesis and Metabolism: Biochemical and Molecular Genetics. Plant Cell 1995, 7, 921–934. [Google Scholar] [CrossRef]
- Ludwig-Müller, J. Auxin conjugates: Their role for plant development and in the evolution of land plants. J. Exp. Bot. 2011, 62, 1757–1773. [Google Scholar] [CrossRef]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-Activated Protein Kinase Cascades in Plant Hormone Signaling. Front. Plant Sci. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Su, V.; Lau, A.F. Ubiquitin-like and ubiquitin-associated domain proteins: Significance in proteasomal degradation. Cell. Mol. Life Sci. 2009, 66, 2819–2833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Liu, H.; Chong, K.; Xu, Y. Roles of ubiquitination-mediated protein degradation in plant responses to abiotic stresses. Environ. Exp. Bot. 2015, 114, 92–103. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2013, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Siedow, J.N.; Umbach, A.L. The mitochondrial cyanideresistant oxidase: Structural conservation amid regulatory diversity. Biochim. Biophys. Acta 2000, 1459, 432–439. [Google Scholar] [CrossRef]
- Djebbar, R.; Rzigui, T.; Pétriacq, P.; Mauve, C.; Priault, P.; Fresneau, C.; De, P.M.; Florez-Sarasa, I.; Benhassaine-Kesri, G.; Streb, P.; et al. Respiratory complex I deficiency induces drought tolerance by impacting leaf stomatal and hydraulic conductances. Planta 2012, 235, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, I.F. Interaction between drought and chronic high temperature during kernel filling in wheat in a controlled environment. Ann. Bot. 2002, 90, 469–476. [Google Scholar] [CrossRef]
- Li, W.; Wei, Z.; Qiao, Z.; Wu, Z.; Cheng, L.; Wang, Y. Proteomics analysis of alfalfa response to heat stress. PLoS ONE 2013, 8, e82725. [Google Scholar] [CrossRef]
- Lee, H.S. Principles and Experimental Techniques of Plant Physiology and Biochemistry, 1st ed.; Higher Education Press: Beijing, China, 2000. (In Chinese) [Google Scholar]
- Han, L.B.; Song, G.L.; Zhang, X. Preliminary observation of physiological responses of three turfgrass species to traffic stress. HortTechnology 2008, 18, 139–143. [Google Scholar] [CrossRef]
- Bates, T.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf senescence: Correlated with increased leaves of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Swägger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, S. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to PEG-Induced Osmotic Stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Accession 1 | Gene Name/ID 2 | Description 3 | Covrg. 4 | Pept. 5 | Log2FC 6 | p Value 7 | Expr. 8 | Pathway 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A0A1D6N230 | Zm00001d042191 | Uncharacterized protein | 11.16 | 6 | 1.234928 | 7.25 × 10−4 | Up | |
| 2 | B6TDF8 | 100282981 | Glyceraldehyde-3-phosphate dehydrogenase | 61.07 | 19 | 1.219132 | 1.36 × 10−4 | Up | Carbon fixation in photosynthetic organisms//Gluconeogenesis |
| 3 | A0A1D6L6C5 | 100284660 | Cytosolic purine 5-nucleotidase | 5.85 | 4 | 1.212466 | 6.72 × 10−4 | Up | |
| 4 | B4FWF5 | 100273587 | Histone deacetylase 6 | 18.38 | 2 | 1.211052 | 1.39 × 10−3 | Up | |
| 5 | B6TPB9 | 100277111 | Pentatricopeptide repeat-containing protein mitochondrial | 16.47 | 6 | 0.824045 | 2.04 × 10−2 | Down | |
| 6 | B4FVQ0 | 100273462 | Pentatricopeptide repeat-containing protein mitochondrial | 4.07 | 2 | 0.798673 | 3.42 × 10−2 | Down | |
| 7 | B6U9Q8 | mTERF family protein | 5.52 | 2 | 0.773541 | 4.68 × 10−2 | Down | ||
| 8 | A0A1D6HT77 | 100283204 | Galactose-1-phosphate uridyl transferase-like protein | 7.34 | 2 | 0.717849 | 1.57 × 10−3 | Down | Galactose metabolism//Amino sugar and nucleotide sugar metabolism |
| No | Accession | Gene Name/ID | Description | Covrg. | Pept. | YE8112 Fold Change | SD_TD Fold Change | Pathway | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Log2FC | p Value | Log2FC | p Value | |||||||
| 1 | B6SHX8 | Uncharacterized protein | 32.43 | 3 | 1.618419 | 1.51 × 10−3 | 0.77178 | 2.65 × 10−2 | ||
| 2 | B6SGF3 | 100280456 | Glyoxalase family protein superfamily | 38.13 | 3 | 0.514965 | 3.06 × 10−2 | 1.912391 | 2.96 × 10−2 | |
| 3 | B4FQG0 | 100282825 | Hydrogen peroxide-induced 1 | 31.67 | 2 | 1.266202 | 4.56 × 10−2 | 0.807211 | 2.67 × 10−2 | |
| 4 | B4FPJ4 | ADP, ATP carrier protein | 13.3 | 8 | 0.668951 | 3.03 × 10−3 | 1.308112 | 2.30 × 10−3 | ||
| 5 | B4FGT5 | P4H7 | Prolyl 4-hydroxylase 7 | 13.76 | 4 | 1.219904 | 4.69 × 10−2 | 0.825568 | 1.46 × 10−2 | Arginine and proline metabolism |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zenda, T.; Liu, S.; Liu, G.; Jin, H.; Dai, L.; Dong, A.; Yang, Y.; Duan, H. Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways. Int. J. Mol. Sci. 2019, 20, 3743. https://doi.org/10.3390/ijms20153743
Wang X, Zenda T, Liu S, Liu G, Jin H, Dai L, Dong A, Yang Y, Duan H. Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways. International Journal of Molecular Sciences. 2019; 20(15):3743. https://doi.org/10.3390/ijms20153743
Chicago/Turabian StyleWang, Xuan, Tinashe Zenda, Songtao Liu, Guo Liu, Hongyu Jin, Liang Dai, Anyi Dong, Yatong Yang, and Huijun Duan. 2019. "Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways" International Journal of Molecular Sciences 20, no. 15: 3743. https://doi.org/10.3390/ijms20153743
APA StyleWang, X., Zenda, T., Liu, S., Liu, G., Jin, H., Dai, L., Dong, A., Yang, Y., & Duan, H. (2019). Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways. International Journal of Molecular Sciences, 20(15), 3743. https://doi.org/10.3390/ijms20153743