Ketogenic Diet in Alzheimer’s Disease



Abstract
:1. Introduction
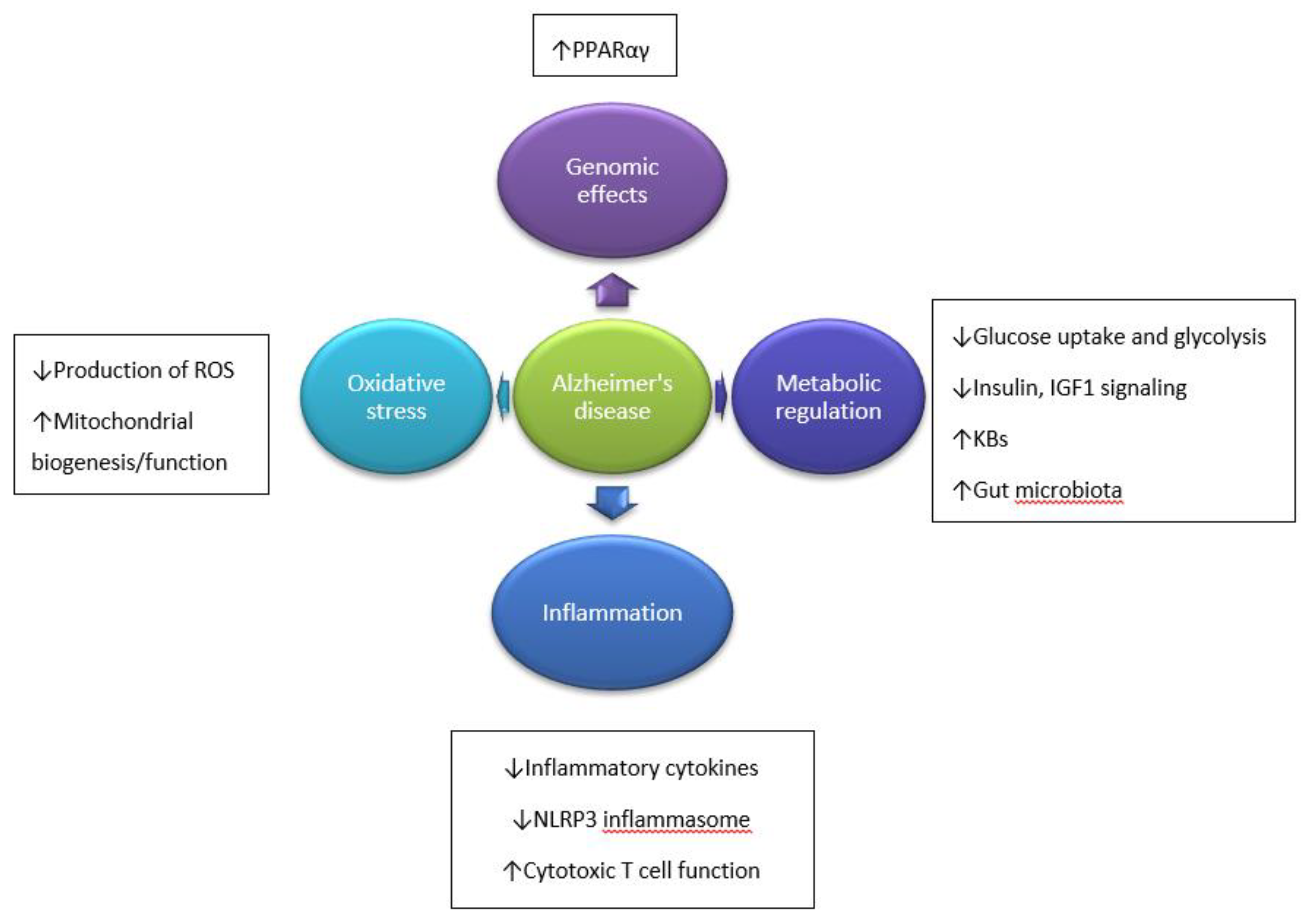
2. Etiopathogenesis of Alzheimer’s Disease
3. Ketogenic Diet
3.1. The Impact of the Ketogenic Diet on Amyloid and Tau Protein
3.2. The Impact of the Ketogenic Diet on Inflammation
3.3. The Impact of the Ketogenic Diet on Dementia
3.4. The Impact of the Ketogenic Diet on Neurodegeneration
3.5. Adverse Effects of the Ketogenic Diet
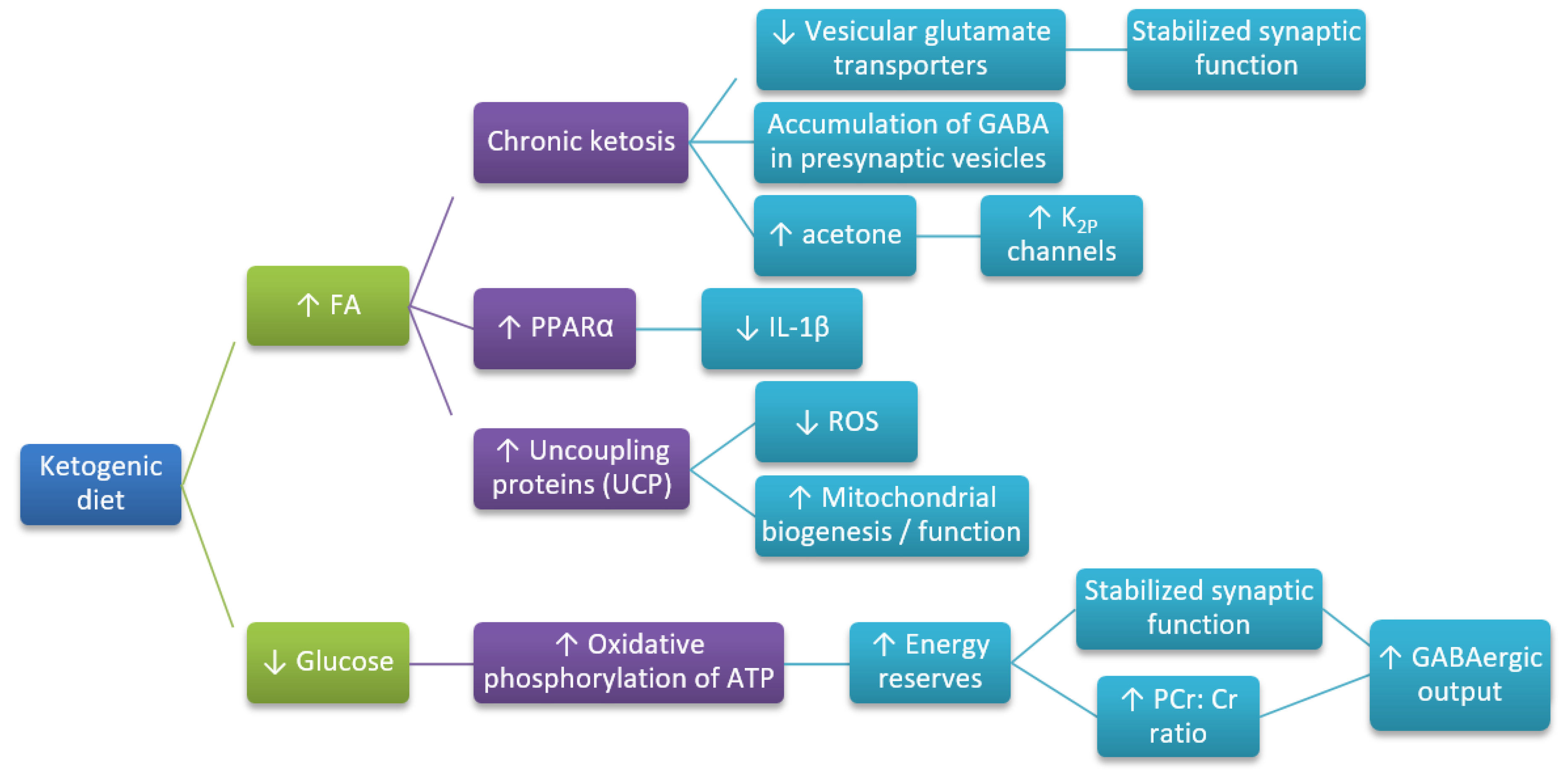
4. The Mechanism of Neuroprotective Action of the Ketogenic Diet
5. Preclinical and Clinical Studies
5.1. Preclinical Studies
5.2. Clinical Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-2-nonenal |
| AcAc | acetoacetate |
| AD | Alzheimer’s disease |
| LD | linear dichroism |
| ADAS-cog | Alzheimer’s Disease Assessment Scale-Cognitive Subscale |
| AGEs | advanced glycation end products |
| AICD | APP intracellular domain |
| ApoE4 | apolipoprotein E4 |
| APP | amyloid precursor protein |
| Aβ | amyloid β-peptide |
| BBB | blood-brain barrier |
| BDH | β-OHB dehydrogenase |
| CNS | central nervous system |
| CR | caloric restriction |
| DASH | Dietary Approaches to Stop Hypertension |
| FAs | fatty acids |
| FIS1 | fission 1 protein (FIS1) |
| FOXO | forkhead transcription factor |
| GABA | γ-aminobutyric acid |
| GSH-Px | glutathione peroxidase |
| GWAS | genome-wide association studies |
| HDACs | histone deacetylases |
| HO-1 | heme oxygenase-1 (HO-1) |
| KBs | ketone bodies |
| KD | ketogenic diet |
| LDL | low-density lipoprotein cholesterol |
| MCI | mild cognitive impairment |
| MCT | medium-chain triglycerides |
| MedDi | Mediterranean diet |
| MIND | Mediterranean-DASH diet Intervention for Neurological Delay |
| mPT | membrane permeability transition |
| mTOR | mammalian target of rapamycin |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFTs | neurofibrillary tangles |
| NLRP3 | NOD-like receptor 3 inflammasome |
| NMDA | N-methyl-D-aspartate |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PCr:Cr | phosphocreatine: Creatine ratio |
| PGC-1α | peroxisome proliferator-activated receptor γ coactivator-1 |
| PHFs | paired helical filaments |
| PPARα | peroxisome proliferator-activated receptor α |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PUFAs | polyunsaturated fatty acids |
| ROS | reactive oxygen species |
| SIRT1 | sirtuin 1 |
| SOD2 | superoxide dismutase 2 |
| UCP | uncoupling proteins |
| β-OHB | β-hydroxybutyrate |
References
- Patterson, C. The World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, September 2018; Available online: https://www.alz.co.uk/research/world-report-2018 (accessed on 5 August 2019).
- Kelley, B.J.; Petersen, R.C. Alzheimer’s disease and mild cognitive impairment. Neurol. Clin. 2007, 25, 577–609. [Google Scholar] [CrossRef]
- Lange, K.W.; Lange, K.M.; Makulska-Gertruda, E.; Nakamura, Y.; Reissmann, A.; Kanaya, S.; Hauser, J. Ketogenic diets and Alzheimer’s disease. Food Sci. Hum. Wellness 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.J.W.; Cervenka, M.C. The expanding role of ketogenic diets in adult neurological disorders. Brain Sci. 2018, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, E.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. (Lond). 2005, 2, 28. [Google Scholar] [CrossRef]
- Beckett, T.L.; Studzinski, C.M.; Keller, J.N.; Paul Murphy, M.; Niedowicz, D.M. A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1505, 61–67. [Google Scholar]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. Plos ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, Q.; Li, S.; Lu, X.; Zhao, Y.; Guan, J.S.; Chen, J.C.; Wu, Q.; Chen, G.Q. 3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer’s disease via mitochondria protection mechanism. Biomaterials 2013, 34, 7552–7562. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Swerdlow, R.H.; Vidoni, E.D.; Morris, J.K.; Mahnken, J.D.; Burns, J.M. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am. J. Clin. Nutr. 2017, 106, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin resistance and neurodegeneration: Progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Raina, P.; Santaguida, P.; Ismaila, A.; Patterson, C.; Cowan, D.; Levine, M.; Booker, L.; Oremus, M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann. Intern. Med. 2008, 148, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Barnard, N.D.; Bush, A.I.; Ceccarelli, A.; Cooper, J.; de Jager, C.A.; Erickson, K.I.; Fraser, G.; Kesler, S.; Levin, S.M.; Lucey, B.; et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 2014, 35, S74–S78. [Google Scholar] [CrossRef]
- Omar, S.H. Mediterranean and MIND diets containing olive biophenols reduces the prevalence of Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 2797. [Google Scholar] [CrossRef]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: New perspectives for neuroprotection in Alzheimer’s disease. Antioxid. (BaselSwitz. ) 2018, 7, 63. [Google Scholar] [CrossRef]
- Huttenlocher, P.R. Ketonemia and seizures: Metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr. Res. 1976, 10, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Zarnowski, T.; Tulidowicz-Bielak, M.; Kosior-Jarecka, E.; Zarnowska, I.A.; Turski, W.; Gasior, M. A ketogenic diet may offer neuroprotection in glaucoma and mitochondrial diseases of the optic nerve. Med. HypothesisDiscov. Innov. Ophthalmol. J. 2012, 1, 45–49. [Google Scholar]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. Bmc Cancer 2008, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. 2), S16. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharm. Exp. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Guerreiro, R.; Hardy, J. Genetics of Alzheimer’s disease. Neurotherapeutics 2014, 11, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar]
- Mondragon-Rodriguez, S.; Perry, G.; Zhu, X.; Moreira, P.I.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell Longev. 2013, 2013, 940603. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Swerdlow, R.H.; Burns, J.M.; Sullivan, D.K. An experimental ketogenic diet for Alzheimer disease was nutritionally dense and rich in vegetables and avocado. Curr. Dev. Nutr. 2019, 3, nzz003. [Google Scholar] [CrossRef]
- Cahill, G.F.J.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A.J.; Kipnis, D.M. Hormone-fuel interrelationships during fasting. J. Clin. Invest. 1966, 45, 1751–1769. [Google Scholar] [CrossRef]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharm.. 2006, 17, 431–439. [Google Scholar] [CrossRef] [Green Version]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef] [Green Version]
- McDonald, T.J.W.; Cervenka, M.C. Ketogenic diets for adult neurological disorders. Neurotherapeutics 2018, 15, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Rho, J.M. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 2007, 48, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Włodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014, 2014, 474296. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F.J. Ketone bodies, potential therapeutic uses. Iubmb Life 2001, 51, 241–247. [Google Scholar] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond) 2009, 6, 31. [Google Scholar] [CrossRef]
- Cahill, G.F.J. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Reynier, P.; Procaccio, V.; Hergesheimer, R.; Corcia, P.; Andres, C.R.; Blasco, H. How can a ketogenic diet improve motor function? Front. Mol. Neurosci. 2018, 11, 15. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. Beta-hydroxybutyrate in the brain: One molecule, multiple mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, G.M.I.; Jiang, L.; Rothman, D.L.; Behar, K.L. The contribution of ketone bodies to basal and activity-dependent deuronal oxidation in vivo. J. Cereb. Blood Flow Metab. 2014, 34, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketone-based metabolic therapy: Is increased NAD+ a primary mechanism? Front. Mol. Neurosci. 2017, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins. Leukot. Essent. Fat. Acids 2004, 70, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Lazarow, A.; Luhovyy, B.; Wehrli, S.; Nissim, I. Response of brain amino acid metabolism to ketosis. Neurochem. Int. 2005, 47, 119–128. [Google Scholar] [CrossRef]
- Yao, J.; Brinton, R.D. Targeting mitochondrial bioenergetics for Alzheimer’s prevention and treatment. Curr. Pharm. Des. 2011, 17, 3474–3479. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.P.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-beta-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.; Curatolo, N.; Benoist, J.F.; Auvin, S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015, 56, e95–e98. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharm. Exp. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado de Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, L.; Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013, 38, 62–76. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. Plos ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.J.; Son, T.G.; Chan, S.L.; Mattson, M.P. Interferon-gamma is up-regulated in the hippocampus in response to intermittent fasting and protects hippocampal neurons against excitotoxicity. J. Neurosci. Res. 2006, 83, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Vanitallie, T.B. Preclinical sporadic Alzheimer’s disease: Target for personalized diagnosis and preventive intervention. Metab. Clin. Exp. 2013, 62, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. Bba Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Ohnuma, T.; Toda, A.; Kimoto, A.; Takebayashi, Y.; Higashiyama, R.; Tagata, Y.; Ito, M.; Ota, T.; Shibata, N.; Arai, H. Benefits of use, and tolerance of, medium-chain triglyceride medical food in the management of Japanese patients with Alzheimer’s disease: A prospective, open-label pilot study. Clin. Interv. Aging 2016, 11, 29–36. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer’s Dement. (N.Y.) 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Dukart, J.; Mueller, K.; Villringer, A.; Kherif, F.; Draganski, B.; Frackowiak, R.; Schroeter, M.L. Relationship between imaging biomarkers, age, progression and symptom severity in Alzheimer’s disease. Neuroimage Clin. 2013, 3, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Courchesne-Loyer, A.; St-Pierre, V.; Vandenberghe, C.; Pierotti, T.; Fortier, M.; Croteau, E.; Castellano, C.A. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2016, 1367, 12–20. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Rettberg, J.R.; Klosinski, L.P.; Cadenas, E.; Brinton, R.D. Shift in brain metabolism in late onset Alzheimer’s disease: Implications for biomarkers and therapeutic interventions. Mol. Asp. Med. 2011, 32, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Hasan-Olive, M.M.; Regnell, C.E.; Kleppa, L.; Scheibye-Knudsen, M.; Gjedde, A.; Klungland, A.; Bohr, V.A.; Storm-Mathisen, J.; Bergersen, L.H. A ketogenic diet accelerates neurodegeneration in mice with induced mitochondrial DNA toxicity in the forebrain. Neurobiol. Aging 2016, 48, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Zelin, E.; Freeman, B.C. Lysine deacetylases regulate the heat shock response including the age-associated impairment of HSF1. J. Mol. Biol. 2015, 427, 1644–1654. [Google Scholar] [CrossRef]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. Plos ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharm.. 2012, 3, 59. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [Green Version]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Jablonski, M. The ketogenic diet for epilepsy therapy in children: Quo vadis? Nutrition 2011, 27, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Ulamek-Koziol, M.; Pluta, R.; Bogucka-Kocka, A.; Czuczwar, S.J. To treat or not to treat drug-refractory epilepsy by the ketogenic diet? That is the question. Ann. Agric. Env. Med. 2016, 23, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.; Janousek, J.; Barber, A.; Weissberger, R. Ketogenic diet treatment in adults with refractory epilepsy. Epilepsy Behav. 2010, 19, 575–579. [Google Scholar] [CrossRef]
- Dashti, H.M.; Mathew, T.C.; Hussein, T.; Asfar, S.K.; Behbahani, A.; Khoursheed, M.A.; Al-Sayer, H.M.; Bo-Abbas, Y.Y.; Al-Zaid, N.S. Long-term effects of a ketogenic diet in obese patients. Exp. Clin. Cardiol. 2004, 9, 200–205. [Google Scholar] [PubMed]
- Masino, S.A.; Rho, J.M. Mechanisms of Ketogenic Diet. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, WA, USA, 2012; pp. 1–28. [Google Scholar]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed]
- Theriault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [PubMed]
- Sah, S.K.; Lee, C.; Jang, J.H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reverse, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Haute, C.V.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Jin, L.W.; Cook, D.G.; Leverenz, J.B.; LeBoeuf, R.C. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport 2002, 13, 455–459. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Holsinger, R.M.D.; McLean, C.A.; Laughton, K.M.; Beyreuther, K.; Evin, G.; Masters, C.L.; Li, Q.X. APP intracellular domain is increased and soluble Abeta is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiol. Dis. 2004, 16, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sun, X.; Eroku, B.O.; Tsipis, C.P.; Puchowicz, M.A.; LaManna, J.C. Diet-induced ketosis improves cognitive performance in aged rats. Adv. Exp. Med. Biol. 2010, 662, 71–75. [Google Scholar] [PubMed]
- Nafar, F.; Clarke, J.P.; Mearow, K.M. Coconut oil protects cortical neurons from amyloid beta toxicity by enhancing signaling of cell survival pathways. Neurochem. Int. 2017, 105, 64–79. [Google Scholar] [CrossRef]
- Studzinski, C.M.; MacKay, W.A.; Beckett, T.L.; Henderson, S.T.; Murphy, M.P.; Sullivan, P.G.; Burnham, W.M. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain Res. 2008, 1226, 209–217. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45 day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Preclinical Studies | ||
|---|---|---|
| Model | Main Findings | Ref. |
| • Hippocampi of juvenile mice | • Improvement of mitochondrial function • Decreased ROS production • Increased cerebral ATP concentrations | [96] |
| • KD in rats | • Reduced insulin levels • Reduced phosphorylation of Akt and S6 • Decreased mTOR activation | [102] |
| • KD in rats | • Increased lipid peroxidation product 4-hydroxy-2-nonenal (4-HNE) levels • Increased activation of Nrf2 | [112] |
| • In vitro models | • Inhibition of histone deacetylases (HDACs) • Increased transcriptional activity of PPAR-γ | [113] |
| • KD in the APP/V717I transgenic mouse model of AD | • Better mitochondrial function • Reduced oxidative stress • Reduced Aβ deposition | [10] |
| • KD in the APP/PS1 mouse model of AD | • Improvement of motor function • Improvement in energy metabolism • Reduced Aβ deposition | [11] [12] |
| • KD in the Tg4510 mouse model of AD | • Improvement of motor function • Improvement in energy metabolism • Reduced Aβ deposition | [13] [14] |
| • Administration of ketone ester in middle-aged mice (8.5 months old) over eight months | • Improvement of cognitive function • Ameliorated Aβ and tau protein pathology | [30] |
| Clinical Evaluation | |||
|---|---|---|---|
| Type of Study | Protocol | Main Findings | Ref. |
| Double-blind placebo-controlled trial | • 20 adult patients with AD or MCI • Administration of MCT | • Significant increases in β-OHB levels moderated by ApoE4 genotype (greater for ApoE4(+) compared to ApoE4(−)) • Improvement of memory and cognitive function in the ADAS-cog test compared to placebo • Patients ApoE4(+) were less responsive to KD compared to ApoE4(−) | [23] |
| Randomized, Double-blind, Placebo-controlled multicenter trial | • 152 adult patients with mild to moderate AD • Administration of AC-1202 over 90 days | • AC-1202 significantly increases a β-OHB level resulted in • Significant improvement in the ADAS-cog test compared to placebo after 45 and 90 days of treatment • Reduced response to AC-1202 in ApoE4(+) patients compared to ApoE4(−) | [47] |
| Other clinical study | • 23 adult patients with MCI • Administration of high carbohydrate or very low carbohydrate diet over six weeks | • Significant improvement in verbal memory performance for the low carbohydrate subjects • Reductions in weight, waist circumference, fasting glucose, and insulin in the low carbohydrate group • KBs levels positively correlated with memory performance | [77] |
| Singe-patient case study | • One adult patient with early-onset AD • Administration of KME over 20 months | • Improved markedly in mood, affect, self-care, and daily activities • Improved cognitive performance • KME-induced hyperketonemia seems robust, convenient, and safe | [78] |
| Pilot and feasibility, randomized, double-blind placebo-controlled parallel trial | • Six adult patients with MCI • Administration of 56 g/day of MCT over 24 weeks | • Increased β-OHB levels • Improvement of memory in mild AD and ApoE4(−) | [79] |
| Prospective, open-label, observational study | • 22 adult patients with mild-to-moderate AD • Administration of a ketogenic meal “Axona” (40 g of powder containing 20 g of caprylic triglycerides) over 90 days | • No improvement in cognitive performance, even in ApoE4(−) patients | [80] |
| Single-arm pilot trial Ketogenic Diet Retention and Feasibility Trial (KDRAFT) | • Fifteen adult patients with mild-to-moderate AD using an MCT-supplemented ≥1:1 ratio KD for three months (a very high-fat ketogenic diet (VHF-KD)) | • Increased β-OHB levels • Improvement in ADAS-cog in 9 out of 10 patients who completed the study and achieved ketosis | [81] |
| Other clinical study | • 19 adult patients • Administration of MCT-supplemented ketogenic meal (Ketonformula®) containing 20 g of MCT | • Increased β-OHB levels •Improvement of cognitive performance • Positive effects on visual attention, working memory, and performing tasks in non-demented patients | [82] |
| Double-blinded, placebo-controlled, randomized clinical trial | • 16 adult patients with mild-to-moderate AD • Administration of caprylidene over 45 days | • Increased cerebral blood flow in patients ApoE4(−) | [114] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. https://doi.org/10.3390/ijms20163892
Rusek M, Pluta R, Ułamek-Kozioł M, Czuczwar SJ. Ketogenic Diet in Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(16):3892. https://doi.org/10.3390/ijms20163892
Chicago/Turabian StyleRusek, Marta, Ryszard Pluta, Marzena Ułamek-Kozioł, and Stanisław J. Czuczwar. 2019. "Ketogenic Diet in Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 16: 3892. https://doi.org/10.3390/ijms20163892
APA StyleRusek, M., Pluta, R., Ułamek-Kozioł, M., & Czuczwar, S. J. (2019). Ketogenic Diet in Alzheimer’s Disease. International Journal of Molecular Sciences, 20(16), 3892. https://doi.org/10.3390/ijms20163892