Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms

 , ,
, , 
Abstract
:1. Introduction
1.1. Somatostatin Receptor Signaling
1.2. Somatostatin Receptor Homo-Dimerization and Hetero-Dimerization
2. “Old” and “New” Somatostatin Receptor Ligands
Differential Functional Properties of Somatostatin Receptor Ligands
3. Comparison between First-Generation and Second-Generation Somatostatin Receptor Ligands in Clinical Studies and Preclinical Models of Pituitary Tumors
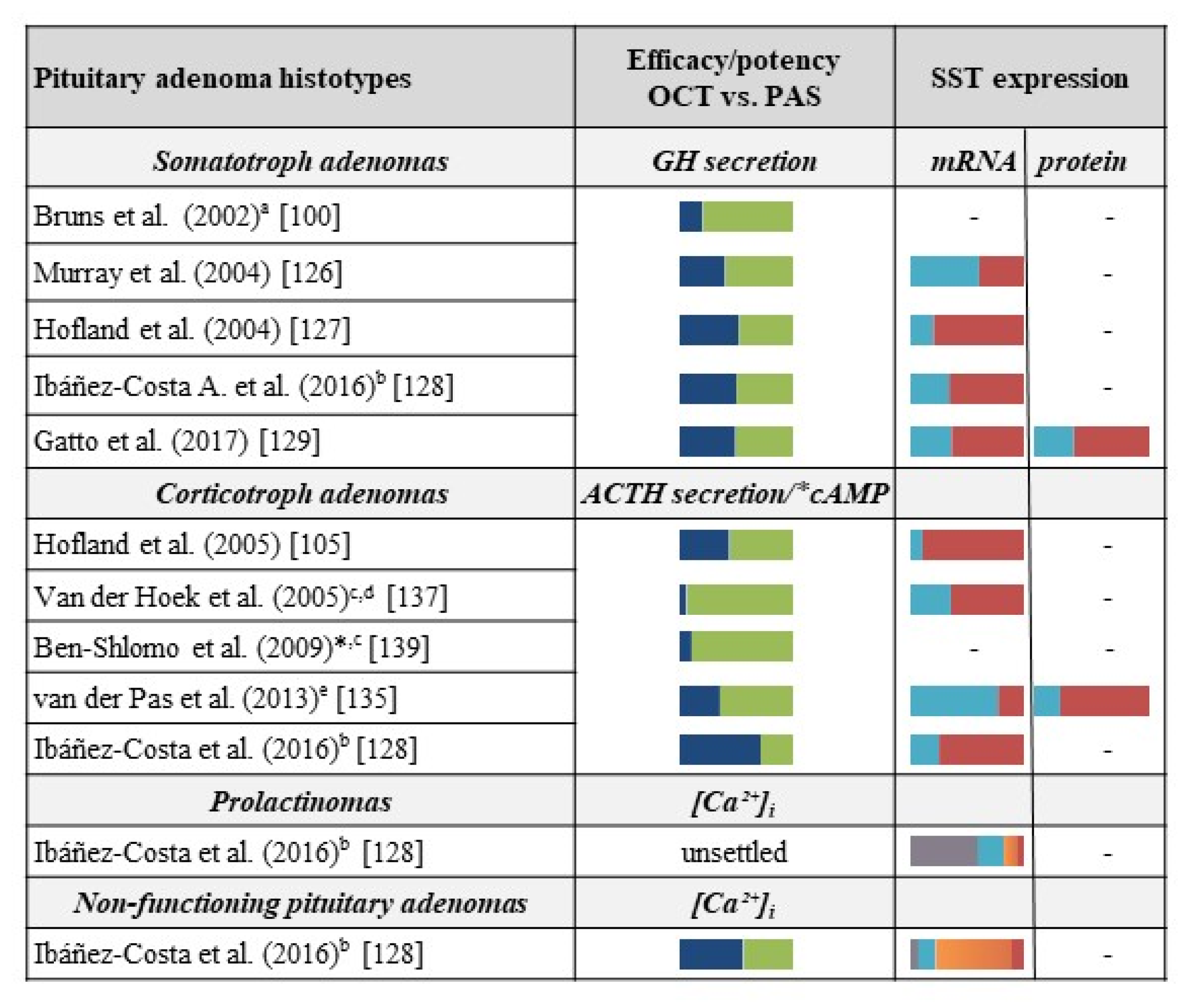
3.1. Acromegaly
3.2. Cushing’s Disease
3.3. Other Pituitary Tumors
4. Comparison between First- and Second−Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms
4.1. Somatostatin Receptor Ligands in Preclinical Models of Neuroendocrine Neoplasms
4.2. Somatostatin Receptor Ligands in Clinical Management of Neuroendocrine Neoplasms: Improvement of Symptoms and Anti-tumor Effects
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Reichlin, S. Somatostatin. New Engl. J. Med. 1983, 309, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castano, J.P.; Wester, H.J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharm. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Wurth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide receptor targeting in cancer: The somatostatin paradigm. Int. J. Pept. 2013, 2013, 926295. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocr. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Ferone, D.; van Hagen, P.M.; van Koetsveld, P.M.; Zuijderwijk, J.; Mooy, D.M.; Lichtenauer-Kaligis, E.G.; Colao, A.; Bogers, A.J.; Lombardi, G.; Lamberts, S.W.; et al. In vitro characterization of somatostatin receptors in the human thymus and effects of somatostatin and octreotide on cultured thymic epithelial cells. Endocrinology 1999, 140, 373–380. [Google Scholar] [CrossRef]
- Ten Bokum, A.M.; Hofland, L.J.; van Hagen, P.M. Somatostatin and somatostatin receptors in the immune system: A review. Eur. Cytokine Netw. 2000, 11, 161–176. [Google Scholar] [PubMed]
- McIntosh, C.H. Gastrointestinal somatostatin: Distribution, secretion and physiological significance. Life Sci. 1985, 37, 2043–2058. [Google Scholar] [CrossRef]
- Artinian, J.; Lacaille, J.C. Disinhibition in learning and memory circuits: New vistas for somatostatin interneurons and long-term synaptic plasticity. Brain Res. Bull. 2018, 141, 20–26. [Google Scholar] [CrossRef]
- Schettini, G.; Florio, T.; Magri, G.; Grimaldi, M.; Meucci, O.; Landolfi, E.; Marino, A. Somatostatin and SMS 201-995 reverse the impairment of cognitive functions induced by cysteamine depletion of brain somatostatin. Eur. J. Pharm. 1988, 151, 399–407. [Google Scholar] [CrossRef]
- Tuboly, G.; Vecsei, L. Somatostatin and cognitive function in neurodegenerative disorders. Mini Rev. Med. Chem. 2013, 13, 34–46. [Google Scholar] [CrossRef]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.M.; Suemoto, T.; Higuchi, M.; Saido, T.C. Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat. Med. 2005, 11, 434–439. [Google Scholar] [CrossRef]
- Schmid, L.C.; Mittag, M.; Poll, S.; Steffen, J.; Wagner, J.; Geis, H.R.; Schwarz, I.; Schmidt, B.; Schwarz, M.K.; Remy, S.; et al. Dysfunction of Somatostatin-Positive Interneurons Associated with Memory Deficits in an Alzheimer’s Disease Model. Neuron 2016, 92, 114–125. [Google Scholar] [CrossRef]
- Shenoy, P.A.; Kuo, A.; Khan, N.; Gorham, L.; Nicholson, J.R.; Corradini, L.; Vetter, I.; Smith, M.T. The Somatostatin Receptor-4 Agonist J-2156 Alleviates Mechanical Hypersensitivity in a Rat Model of Breast Cancer Induced Bone Pain. Front. Pharm. 2018, 9, 495. [Google Scholar] [CrossRef]
- Riedemann, T. Diversity and Function of Somatostatin-Expressing Interneurons in the Cerebral Cortex. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- De Lecea, L. Cortistatin--functions in the central nervous system. Mol. Cell. Endocrinol. 2008, 286, 88–95. [Google Scholar] [CrossRef]
- Ruscica, M.; Arvigo, M.; Steffani, L.; Ferone, D.; Magni, P. Somatostatin, somatostatin analogs and somatostatin receptor dynamics in the biology of cancer progression. Curr. Mol. Med. 2013, 13, 555–571. [Google Scholar] [CrossRef]
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973, 179, 77–79. [Google Scholar] [CrossRef]
- Bauer, W.; Briner, U.; Doepfner, W.; Haller, R.; Huguenin, R.; Marbach, P.; Petcher, T.J. SMS 201-995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Barbieri, F.; Albertelli, M.; Grillo, F.; Mohamed, A.; Saveanu, A.; Barlier, A.; Ferone, D.; Florio, T. Neuroendocrine tumors: Insights into innovative therapeutic options and rational development of targeted therapies. Drug Discov. Today 2014, 19, 458–468. [Google Scholar] [CrossRef]
- Florio, T. Molecular mechanisms of the antiproliferative activity of somatostatin receptors (SSTRs) in neuroendocrine tumors. Front. Biosci. A J. Virtual Libr. 2008, 13, 822–840. [Google Scholar] [CrossRef]
- Ben-Shlomo, A.; Liu, N.A.; Melmed, S. Somatostatin and dopamine receptor regulation of pituitary somatotroph adenomas. Pituitary 2017, 20, 93–99. [Google Scholar] [CrossRef]
- Merola, E.; Panzuto, F.; Delle Fave, G. Antiproliferative effect of somatostatin analogs in advanced gastro-entero-pancreatic neuroendocrine tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 46624–46634. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Cwikla, J.B.; Phan, A.T.; Raderer, M.; Sedlackova, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. New Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Rinke, A.; Muller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Blaker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef]
- Hasskarl, J.; Kaufmann, M.; Schmid, H.A. Somatostatin receptors in non-neuroendocrine malignancies: The potential role of somatostatin analogs in solid tumors. Future Oncol. 2011, 7, 895–913. [Google Scholar] [CrossRef]
- Keskin, O.; Yalcin, S. A review of the use of somatostatin analogs in oncology. OncoTargets Ther. 2013, 6, 471–483. [Google Scholar] [Green Version]
- Michael, M.; Garcia-Carbonero, R.; Weber, M.M.; Lombard-Bohas, C.; Toumpanakis, C.; Hicks, R.J. The Antiproliferative Role of Lanreotide in Controlling Growth of Neuroendocrine Tumors: A Systematic Review. Oncologist 2017, 22, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Smitha, M.C.; Maggi, M.; Orlando, C. Somatostatin receptors in non-endocrine tumours. Dig. Liver Dis. 2004, 36, S78–S85. [Google Scholar] [CrossRef]
- Law, S.F.; Yasuda, K.; Bell, G.I.; Reisine, T. Gi alpha 3 and G (o) alpha selectively associate with the cloned somatostatin receptor subtype SSTR2. J. Biol. Chem. 1993, 268, 10721–10727. [Google Scholar]
- Hershberger, R.E.; Newman, B.L.; Florio, T.; Bunzow, J.; Civelli, O.; Li, X.J.; Forte, M.; Stork, P.J. The somatostatin receptors SSTR1 and SSTR2 are coupled to inhibition of adenylyl cyclase in Chinese hamster ovary cells via pertussis toxin-sensitive pathways. Endocrinology 1994, 134, 1277–1285. [Google Scholar] [CrossRef]
- Patel, Y.C.; Greenwood, M.T.; Warszynska, A.; Panetta, R.; Srikant, C.B. All five cloned human somatostatin receptors (hSSTR1-5) are functionally coupled to adenylyl cyclase. Biochem. Biophys. Res. Commun. 1994, 198, 605–612. [Google Scholar] [CrossRef]
- Yasuda, K.; Rens-Domiano, S.; Breder, C.D.; Law, S.F.; Saper, C.B.; Reisine, T.; Bell, G.I. Cloning of a novel somatostatin receptor, SSTR3, coupled to adenylylcyclase. J. Biol. Chem. 1992, 267, 20422–20428. [Google Scholar]
- Scherubl, H.; Hescheler, J.; Riecken, E.O. Molecular mechanisms of somatostatin’s inhibition of hormone release: Participation of voltage-gated calcium channels and G-proteins. Horm. Metab. Res. Suppl. Ser. 1993, 27, 1–4. [Google Scholar]
- Yatani, A.; Birnbaumer, L.; Brown, A.M. Direct coupling of the somatostatin receptor to potassium channels by a G protein. Metab. Clin. Exp. 1990, 39, 91–95. [Google Scholar] [CrossRef]
- Schettini, G.; Florio, T.; Meucci, O.; Landolfi, E.; Grimaldi, M.; Ventra, C.; Marino, A. Somatostatin inhibition of adenylate cyclase activity in different brain areas. Brain Res. 1989, 492, 65–71. [Google Scholar] [CrossRef]
- Charland, S.; Boucher, M.J.; Houde, M.; Rivard, N. Somatostatin inhibits Akt phosphorylation and cell cycle entry, but not p42/p44 mitogen-activated protein (MAP) kinase activation in normal and tumoral pancreatic acinar cells. Endocrinology 2001, 142121–142128. [Google Scholar] [CrossRef]
- Florio, T.; Yao, H.; Carey, K.D.; Dillon, T.J.; Stork, P.J. Somatostatin activation of mitogen-activated protein kinase via somatostatin receptor 1 (SSTR1). Mol. Endocrinol. 1999, 13, 24–37. [Google Scholar] [CrossRef]
- Pages, P.; Benali, N.; Saint-Laurent, N.; Esteve, J.P.; Schally, A.V.; Tkaczuk, J.; Vaysse, N.; Susini, C.; Buscail, L. sst2 somatostatin receptor mediates cell cycle arrest and induction of p27(Kip1). Evidence for the role of SHP-1. J. Biol. Chem. 1999, 274, 15186–15193. [Google Scholar] [CrossRef]
- Ruscica, M.; Magni, P.; Steffani, L.; Gatto, F.; Albertelli, M.; Rametta, R.; Valenti, L.; Ameri, P.; Magnaghi, V.; Culler, M.D.; et al. Characterization and sub-cellular localization of SS1R, SS2R, and SS5R in human late-stage prostate cancer cells: Effect of mono- and bi-specific somatostatin analogs on cell growth. Mol. Cell. Endocrinol. 2014, 382, 860–870. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Zhang, J.; Laupheimer, S.; Paez-Pereda, M.; Erneux, C.; Florio, T.; Pagotto, U.; Stalla, G.K. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res. 2006, 66, 1576–1582. [Google Scholar] [CrossRef]
- Barbieri, F.; Pattarozzi, A.; Gatti, M.; Porcile, C.; Bajetto, A.; Ferrari, A.; Culler, M.D.; Florio, T. Somatostatin receptors 1, 2, and 5 cooperate in the somatostatin inhibition of C6 glioma cell proliferation in vitro via a phosphotyrosine phosphatase-eta-dependent inhibition of extracellularly regulated kinase-1/2. Endocrinology 2008, 149, 4736–4746. [Google Scholar] [CrossRef]
- Cordelier, P.; Esteve, J.P.; Bousquet, C.; Delesque, N.; O’Carroll, A.M.; Schally, A.V.; Vaysse, N.; Susini, C.; Buscail, L. Characterization of the antiproliferative signal mediated by the somatostatin receptor subtype sst5. Proc. Natl. Acad. Sci. USA 1997, 94, 9343–9348. [Google Scholar] [CrossRef] [Green Version]
- Hubina, E.; Nanzer, A.M.; Hanson, M.R.; Ciccarelli, E.; Losa, M.; Gaia, D.; Papotti, M.; Terreni, M.R.; Khalaf, S.; Jordan, S.; et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur. J. Endocrinol. 2006, 155, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Lahlou, H.; Saint-Laurent, N.; Esteve, J.P.; Eychene, A.; Pradayrol, L.; Pyronnet, S.; Susini, C. sst2 Somatostatin receptor inhibits cell proliferation through Ras-, Rap1-, and B-Raf-dependent ERK2 activation. J. Biol. Chem. 2003, 278, 39356–39371. [Google Scholar] [CrossRef]
- Pola, S.; Cattaneo, M.G.; Vicentini, L.M. Anti-migratory and anti-invasive effect of somatostatin in human neuroblastoma cells: Involvement of Rac and MAP kinase activity. J. Biol. Chem. 2003, 278, 40601–40606. [Google Scholar] [CrossRef]
- Pan, M.G.; Florio, T.; Stork, P.J. G protein activation of a hormone-stimulated phosphatase in human tumor cells. Science 1992, 256, 1215–1217. [Google Scholar] [CrossRef]
- Florio, T. Somatostatin/somatostatin receptor signalling: Phosphotyrosine phosphatases. Mol. Cell. Endocrinol. 2008, 286, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Florio, T.; Arena, S.; Thellung, S.; Iuliano, R.; Corsaro, A.; Massa, A.; Pattarozzi, A.; Bajetto, A.; Trapasso, F.; Fusco, A.; et al. The activation of the phosphotyrosine phosphatase eta (r-PTP eta) is responsible for the somatostatin inhibition of PC Cl3 thyroid cell proliferation. Mol. Endocrinol. 2001, 15, 1838–1852. [Google Scholar]
- Florio, T.; Thellung, S.; Arena, S.; Corsaro, A.; Spaziante, R.; Gussoni, G.; Acuto, G.; Giusti, M.; Giordano, G.; Schettini, G. Somatostatin and its analog lanreotide inhibit the proliferation of dispersed human non-functioning pituitary adenoma cells in vitro. Eur. J. Endocrinol. 1999, 141, 396–408. [Google Scholar] [CrossRef]
- Florio, T.; Thellung, S.; Corsaro, A.; Bocca, L.; Arena, S.; Pattarozzi, A.; Villa, V.; Massa, A.; Diana, F.; Schettini, D.; et al. Characterization of the intracellular mechanisms mediating somatostatin and lanreotide inhibition of DNA synthesis and growth hormone release from dispersed human GH-secreting pituitary adenoma cells in vitro. Clin. Endocrinol. 2003, 59, 115–128. [Google Scholar] [CrossRef]
- Cerovac, V.; Monteserin-Garcia, J.; Rubinfeld, H.; Buchfelder, M.; Losa, M.; Florio, T.; Paez-Pereda, M.; Stalla, G.K.; Theodoropoulou, M. The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res. 2010, 70, 666–674. [Google Scholar] [CrossRef]
- Florio, T.; Rim, C.; Hershberger, R.E.; Loda, M.; Stork, P.J. The somatostatin receptor SSTR1 is coupled to phosphotyrosine phosphatase activity in CHO-K1 cells. Mol. Endocrinol. 1994, 8, 1289–1297. [Google Scholar]
- Florio, T.; Thellung, S.; Arena, S.; Corsaro, A.; Bajetto, A.; Schettini, G.; Stork, P.J. Somatostatin receptor 1 (SSTR1)-mediated inhibition of cell proliferation correlates with the activation of the MAP kinase cascade: Role of the phosphotyrosine phosphatase SHP-2. J. Physiol. Paris 2000, 94, 239–250. [Google Scholar] [CrossRef]
- Lopez, F.; Esteve, J.P.; Buscail, L.; Delesque, N.; Saint-Laurent, N.; Theveniau, M.; Nahmias, C.; Vaysse, N.; Susini, C. The tyrosine phosphatase SHP-1 associates with the sst2 somatostatin receptor and is an essential component of sst2-mediated inhibitory growth signaling. J. Biol. Chem. 1997, 272, 24448–24454. [Google Scholar] [CrossRef]
- Reardon, D.B.; Dent, P.; Wood, S.L.; Kong, T.; Sturgill, T.W. Activation in vitro of somatostatin receptor subtypes 2, 3, or 4 stimulates protein tyrosine phosphatase activity in membranes from transfected Ras-transformed NIH 3T3 cells: Coexpression with catalytically inactive SHP-2 blocks responsiveness. Mol. Endocrinol. 1997, 11, 1062–1069. [Google Scholar] [CrossRef]
- Yamamoto, J.; Ohnuma, K.; Hatano, R.; Okamoto, T.; Komiya, E.; Yamazaki, H.; Iwata, S.; Dang, N.H.; Aoe, K.; Kishimoto, T.; et al. Regulation of somatostatin receptor 4-mediated cytostatic effects by CD26 in malignant pleural mesothelioma. Br. J. Cancer 2014, 110, 2232–2245. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, F.; Pattarozzi, A.; Gatti, M.; Aiello, C.; Quintero, A.; Lunardi, G.; Bajetto, A.; Ferrari, A.; Culler, M.D.; Florio, T. Differential efficacy of SSTR1, -2, and -5 agonists in the inhibition of C6 glioma growth in nude mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1078–E1088. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Taylor, J.E.; Culler, M.D.; Nisoli, E.; Vicentini, L.M. Selective stimulation of somatostatin receptor subtypes: Differential effects on Ras/MAP kinase pathway and cell proliferation in human neuroblastoma cells. FEBS Lett. 2000, 481, 271–276. [Google Scholar] [CrossRef]
- Akbar, M.; Okajima, F.; Tomura, H.; Majid, M.A.; Yamada, Y.; Seino, S.; Kondo, Y. Phospholipase C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1-5, in transfected COS-7 cells. FEBS Lett. 1994, 348, 192–196. [Google Scholar] [CrossRef]
- Kim, J.K.; Kwon, O.; Kim, J.; Kim, E.K.; Park, H.K.; Lee, J.E.; Kim, K.L.; Choi, J.W.; Lim, S.; Seok, H.; et al. PDZ domain-containing 1 (PDZK1) protein regulates phospholipase C-beta3 (PLC-beta3)-specific activation of somatostatin by forming a ternary complex with PLC-beta3 and somatostatin receptors. J. Biol. Chem. 2012, 287, 21012–21024. [Google Scholar] [CrossRef]
- Alderton, F.; Humphrey, P.P.; Sellers, L.A. High-intensity p38 kinase activity is critical for p21 (cip1) induction and the antiproliferative function of G (i) protein-coupled receptors. Mol. Pharmacol. 2001, 59, 1119–1128. [Google Scholar] [CrossRef]
- War, S.A.; Kim, B.; Kumar, U. Human somatostatin receptor-3 distinctively induces apoptosis in MCF-7 and cell cycle arrest in MDA-MB-231 breast cancer cells. Mol. Cell. Endocrinol. 2015, 413, 129–144. [Google Scholar] [CrossRef]
- Sharma, K.; Patel, Y.C.; Srikant, C.B. Subtype-selective induction of wild-type p53 and apoptosis, but not cell cycle arrest, by human somatostatin receptor 3. Mol. Endocrinol. 1996, 10, 1688–1696. [Google Scholar]
- Guillermet, J.; Saint-Laurent, N.; Rochaix, P.; Cuvillier, O.; Levade, T.; Schally, A.V.; Pradayrol, L.; Buscail, L.; Susini, C.; Bousquet, C. Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 155–160. [Google Scholar] [CrossRef]
- Scoazec, J.Y. Angiogenesis in neuroendocrine tumors: Therapeutic applications. Neuroendocrinology 2013, 97, 45–56. [Google Scholar] [CrossRef]
- Albini, A.; Florio, T.; Giunciuglio, D.; Masiello, L.; Carlone, S.; Corsaro, A.; Thellung, S.; Cai, T.; Noonan, D.M.; Schettini, G. Somatostatin controls Kaposi’s sarcoma tumor growth through inhibition of angiogenesis. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 647–655. [Google Scholar] [CrossRef]
- Bocci, G.; Culler, M.D.; Fioravanti, A.; Orlandi, P.; Fasciani, A.; Colucci, R.; Taylor, J.E.; Sadat, D.; Danesi, R.; Del Tacca, M. In vitro antiangiogenic activity of selective somatostatin subtype-1 receptor agonists. Eur. J. Clin. Investig. 2007, 37, 700–708. [Google Scholar] [CrossRef]
- Kumar, M.; Liu, Z.R.; Thapa, L.; Chang, Q.; Wang, D.Y.; Qin, R.Y. Antiangiogenic effect of somatostatin receptor subtype 2 on pancreatic cancer cell line: Inhibition of vascular endothelial growth factor and matrix metalloproteinase-2 expression in vitro. World J. Gastroenterol 2004, 10, 393–399. [Google Scholar] [CrossRef]
- Florio, T.; Morini, M.; Villa, V.; Arena, S.; Corsaro, A.; Thellung, S.; Culler, M.D.; Pfeffer, U.; Noonan, D.M.; Schettini, G.; et al. Somatostatin inhibits tumor angiogenesis and growth via somatostatin receptor-3-mediated regulation of endothelial nitric oxide synthase and mitogen-activated protein kinase activities. Endocrinology 2003, 144, 1574–1584. [Google Scholar] [CrossRef]
- Patel, P.C.; Barrie, R.; Hill, N.; Landeck, S.; Kurozawa, D.; Woltering, E.A. Postreceptor signal transduction mechanisms involved in octreotide-induced inhibition of angiogenesis. Surgery 1994, 116, 1148–1152. [Google Scholar]
- Arena, S.; Pattarozzi, A.; Corsaro, A.; Schettini, G.; Florio, T. Somatostatin receptor subtype-dependent regulation of nitric oxide release: Involvement of different intracellular pathways. Mol. Endocrinol. 2005, 19, 255–267. [Google Scholar] [CrossRef]
- Cordelier, P.; Esteve, J.P.; Najib, S.; Moroder, L.; Vaysse, N.; Pradayrol, L.; Susini, C.; Buscail, L. Regulation of neuronal nitric-oxide synthase activity by somatostatin analogs following SST5 somatostatin receptor activation. J. Biol. Chem. 2006, 281, 19156–19171. [Google Scholar] [CrossRef]
- Cordoba-Chacon, J.; Gahete, M.D.; Duran-Prado, M.; Luque, R.M.; Castano, J.P. Truncated somatostatin receptors as new players in somatostatin-cortistatin pathophysiology. Ann. New York Acad. Sci. 2011, 1220, 6–15. [Google Scholar] [CrossRef]
- Gahete, M.D.; Rincon-Fernandez, D.; Duran-Prado, M.; Hergueta-Redondo, M.; Ibanez-Costa, A.; Rojo-Sebastian, A.; Gracia-Navarro, F.; Culler, M.D.; Casanovas, O.; Moreno-Bueno, G.; et al. The truncated somatostatin receptor sst5TMD4 stimulates the angiogenic process and is associated to lymphatic metastasis and disease-free survival in breast cancer patients. Oncotarget 2016, 7, 60110–60122. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Varma, M.G.; Joubel, A.; Madabushi, S.; Lichtarge, O.; Barber, D.L. Conserved motifs in somatostatin, D2-dopamine, and alpha 2B-adrenergic receptors for inhibiting the Na-H exchanger, NHE1. J. Biol. Chem. 2003, 278, 15128–15135. [Google Scholar] [CrossRef]
- Buchan, A.M.; Lin, C.Y.; Choi, J.; Barber, D.L. Somatostatin, acting at receptor subtype 1, inhibits Rho activity, the assembly of actin stress fibers, and cell migration. J. Biol. Chem. 2002, 277, 28431–28438. [Google Scholar] [CrossRef]
- Bulenger, S.; Marullo, S.; Bouvier, M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 2005, 26, 131–137. [Google Scholar] [CrossRef]
- Milligan, G. G protein-coupled receptor dimerisation: Molecular basis and relevance to function. Biochim. Et Biophys. Acta 2007, 1768, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Grant, M.; Collier, B.; Kumar, U. Agonist-dependent dissociation of human somatostatin receptor 2 dimers: A role in receptor trafficking. J. Biol. Chem. 2004, 279, 36179–36183. [Google Scholar] [CrossRef]
- Pfeiffer, M.; Koch, T.; Schroder, H.; Klutzny, M.; Kirscht, S.; Kreienkamp, H.J.; Hollt, V.; Schulz, S. Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst (3) receptor function by heterodimerization with sst (2A). J. Biol. Chem. 2001, 276, 14027–14036. [Google Scholar] [CrossRef]
- Grant, M.; Patel, R.C.; Kumar, U. The role of subtype-specific ligand binding and the C-tail domain in dimer formation of human somatostatin receptors. J. Biol. Chem. 2004, 279, 38636–38643. [Google Scholar] [CrossRef]
- Patel, R.C.; Kumar, U.; Lamb, D.C.; Eid, J.S.; Rocheville, M.; Grant, M.; Rani, A.; Hazlett, T.; Patel, S.C.; Gratton, E.; et al. Ligand binding to somatostatin receptors induces receptor-specific oligomer formation in live cells. Proc. Natl. Acad. Sci. USA. 2002, 99, 3294–3299. [Google Scholar] [CrossRef] [Green Version]
- Rocheville, M.; Lange, D.C.; Kumar, U.; Sasi, R.; Patel, R.C.; Patel, Y.C. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J. Biol. Chem. 2000, 275, 7862–7869. [Google Scholar] [CrossRef]
- Ruscica, M.; Arvigo, M.; Gatto, F.; Dozio, E.; Feltrin, D.; Culler, M.D.; Minuto, F.; Motta, M.; Ferone, D.; Magni, P. Regulation of prostate cancer cell proliferation by somatostatin receptor activation. Mol. Cell. Endocrinol. 2010, 315, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Tan, H.; Zhao, Y.; Zhou, Y.; Cao, L. Expression and selective activation of somatostatin receptor subtypes induces cell cycle arrest in cancer cells. Oncol. Lett. 2019, 17, 1723–1731. [Google Scholar] [CrossRef]
- Grant, M.; Alturaihi, H.; Jaquet, P.; Collier, B.; Kumar, U. Cell growth inhibition and functioning of human somatostatin receptor type 2 are modulated by receptor heterodimerization. Mol. Endocrinol. 2008, 22, 2278–2292. [Google Scholar] [CrossRef]
- Pfeiffer, M.; Koch, T.; Schroder, H.; Laugsch, M.; Hollt, V.; Schulz, S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J. Biol. Chem. 2002, 277, 19762–19772. [Google Scholar] [CrossRef]
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for dopamine and somatostatin: Formation of hetero-oligomers with enhanced functional activity. Science 2000, 288, 154–157. [Google Scholar] [CrossRef]
- Baragli, A.; Alturaihi, H.; Watt, H.L.; Abdallah, A.; Kumar, U. Heterooligomerization of human dopamine receptor 2 and somatostatin receptor 2 Co-immunoprecipitation and fluorescence resonance energy transfer analysis. Cell. Signal. 2007, 19, 2304–2316. [Google Scholar] [CrossRef]
- Somvanshi, R.K.; War, S.A.; Chaudhari, N.; Qiu, X.; Kumar, U. Receptor specific crosstalk and modulation of signaling upon heterodimerization between beta1-adrenergic receptor and somatostatin receptor-5. Cell. Signal. 2011, 23, 794–811. [Google Scholar] [CrossRef]
- Taylor, J.E.; Bogden, A.E.; Moreau, J.P.; Coy, D.H. In vitro and in vivo inhibition of human small cell lung carcinoma (NCI-H69) growth by a somatostatin analogue. Biochem. Biophys. Res. Commun. 1988, 153, 81–86. [Google Scholar] [CrossRef]
- Kuhn, J.M.; Basin, C.; Mollard, M.; de Rouge, B.; Baudoin, C.; Obach, R.; Tolis, G. Pharmacokinetic study and effects on growth hormone secretion in healthy volunteers of the new somatostatin analogue BIM 23014. Eur. J. Clin. Pharm. 1993, 45, 73–77. [Google Scholar] [CrossRef]
- Ben-Shlomo, A.; Melmed, S. Somatostatin agonists for treatment of acromegaly. Mol. Cell. Endocrinol. 2008, 286, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Gadelha, M.R.; Wildemberg, L.E.; Bronstein, M.D.; Gatto, F.; Ferone, D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 2017, 20, 100–108. [Google Scholar] [CrossRef]
- Oberg, K.; Lamberts, S.W. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: Past, present and future. Endocr. -Relat. Cancer 2016, 23, R551–R566. [Google Scholar] [CrossRef]
- Lancranjan, I.; Bruns, C.; Grass, P.; Jaquet, P.; Jervell, J.; Kendall-Taylor, P.; Lamberts, S.W.; Marbach, P.; Orskov, H.; Pagani, G.; et al. Sandostatin LAR: A promising therapeutic tool in the management of acromegalic patients. Metabolism 1996, 45, 67–71. [Google Scholar] [CrossRef]
- Pouget, E.; Fay, N.; Dujardin, E.; Jamin, N.; Berthault, P.; Perrin, L.; Pandit, A.; Rose, T.; Valery, C.; Thomas, D.; et al. Elucidation of the self-assembly pathway of lanreotide octapeptide into beta-sheet nanotubes: Role of two stable intermediates. J. Am. Chem. Soc. 2010, 132, 4230–4241. [Google Scholar] [CrossRef]
- Caron, P.; Beckers, A.; Cullen, D.R.; Goth, M.I.; Gutt, B.; Laurberg, P.; Pico, A.M.; Valimaki, M.; Zgliczynski, W. Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in the management of acromegaly. J. Clin. Endocrinol. Metab. 2002, 87, 99–104. [Google Scholar] [CrossRef]
- Astruc, B.; Marbach, P.; Bouterfa, H.; Denot, C.; Safari, M.; Vitaliti, A.; Sheppard, M. Long-acting octreotide and prolonged-release lanreotide formulations have different pharmacokinetic profiles. J. Clin. Pharmacol. 2005, 45, 836–844. [Google Scholar] [CrossRef]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef]
- Reubi, J.C.; Eisenwiener, K.P.; Rink, H.; Waser, B.; Macke, H.R. A new peptidic somatostatin agonist with high affinity to all five somatostatin receptors. Eur. J. Pharm. 2002, 456, 45–49. [Google Scholar] [CrossRef]
- Petersenn, S.; Hu, K.; Maldonado, M.; Zhang, Y.; Lasher, J.; Bouillaud, E.; Wang, Y.; Mann, K.; Unger, N. Tolerability and dose proportional pharmacokinetics of pasireotide administered as a single dose or two divided doses in healthy male volunteers: A single-center, open-label, ascending-dose study. Clin. Ther. 2012, 34, 677–688. [Google Scholar] [CrossRef]
- Dietrich, H.; Hu, K.; Ruffin, M.; Song, D.; Bouillaud, E.; Wang, Y.; Hasskarl, J. Safety, tolerability, and pharmacokinetics of a single dose of pasireotide long-acting release in healthy volunteers: A single-center Phase I study. Eur. J. Endocrinol. 2012, 166, 821–828. [Google Scholar] [CrossRef]
- Lesche, S.; Lehmann, D.; Nagel, F.; Schmid, H.A.; Schulz, S. Differential effects of octreotide and pasireotide on somatostatin receptor internalization and trafficking in vitro. J. Clin. Endocrinol. Metab. 2009, 94, 654–661. [Google Scholar] [CrossRef]
- Hofland, L.J.; van der Hoek, J.; Feelders, R.; van Aken, M.O.; van Koetsveld, P.M.; Waaijers, M.; Sprij-Mooij, D.; Bruns, C.; Weckbecker, G.; de Herder, W.W.; et al. The multi-ligand somatostatin analogue SOM230 inhibits ACTH secretion by cultured human corticotroph adenomas via somatostatin receptor type 5. Eur. J. Endocrinol. 2005, 152, 645–654. [Google Scholar] [CrossRef]
- Gatto, F.; Arvigo, M.; Amaru, J.; Campana, C.; Cocchiara, F.; Graziani, G.; Bruzzone, E.; Giusti, M.; Boschetti, M.; Ferone, D. Cell specific interaction of pasireotide: Review of preclinical studies in somatotroph and corticotroph pituitary cells. Pituitary 2019, 22, 89–99. [Google Scholar] [CrossRef]
- Lahlou, H.; Guillermet, J.; Hortala, M.; Vernejoul, F.; Pyronnet, S.; Bousquet, C.; Susini, C. Molecular signaling of somatostatin receptors. Ann. New York Acad. Sci. 2004, 1014, 121–131. [Google Scholar] [CrossRef]
- Schonbrunn, A. Selective agonism in somatostatin receptor signaling and regulation. Mol. Cell. Endocrinol. 2008, 286, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.J.; Ghosh, M.; Schonbrunn, A. Ligand-dependent mechanisms of sst2A receptor trafficking: Role of site-specific phosphorylation and receptor activation in the actions of biased somatostatin agonists. Mol. Endocrinol. 2011, 25, 1040–1054. [Google Scholar] [CrossRef]
- Poll, F.; Lehmann, D.; Illing, S.; Ginj, M.; Jacobs, S.; Lupp, A.; Stumm, R.; Schulz, S. Pasireotide and octreotide stimulate distinct patterns of sst2A somatostatin receptor phosphorylation. Mol. Endocrinol. 2010, 24, 436–446. [Google Scholar] [CrossRef]
- Cescato, R.; Loesch, K.A.; Waser, B.; Macke, H.R.; Rivier, J.E.; Reubi, J.C.; Schonbrunn, A. Agonist-biased signaling at the sst2A receptor: The multi-somatostatin analogs KE108 and SOM230 activate and antagonize distinct signaling pathways. Mol. Endocrinol. 2010, 24, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Nagel, F.; Doll, C.; Poll, F.; Kliewer, A.; Schroder, H.; Schulz, S. Structural determinants of agonist-selective signaling at the sst (2A) somatostatin receptor. Mol. Endocrinol. 2011, 25, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharm. Ther. 2006, 110, 465–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, R.H.; Revollo, J.; Cidlowski, J.A. Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 17591–17596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Bee, M.S.; Schonbrunn, A. Site specificity of agonist and second messenger-activated kinases for somatostatin receptor subtype 2A (Sst2A) phosphorylation. Mol. Pharmacol. 2009, 76, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Kliewer, A.; Schutz, D.; Nagel, F.; Stumm, R.; Schulz, S. Carboxyl-terminal multi-site phosphorylation regulates internalization and desensitization of the human sst2 somatostatin receptor. Mol. Cell. Endocrinol. 2014, 387, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Blanchard, M.P.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Monges, G.; Garcia, S.; Ferone, D.; et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in human pancreatic neuroendocrine tumor cultures. Endocr. Relat. Cancer 2014, 21, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Gatto, F.; Biermasz, N.R.; Feelders, R.A.; Kros, J.M.; Dogan, F.; van der Lely, A.J.; Neggers, S.J.; Lamberts, S.W.; Pereira, A.M.; Ferone, D.; et al. Low beta-arrestin expression correlates with the responsiveness to long-term somatostatin analog treatment in acromegaly. Eur. J. Endocrinol. 2016, 174, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Gatto, F.; Feelders, R.; van der Pas, R.; Kros, J.M.; Dogan, F.; van Koetsveld, P.M.; van der Lelij, A.J.; Neggers, S.J.; Minuto, F.; de Herder, W.; et al. beta-Arrestin 1 and 2 and G protein-coupled receptor kinase 2 expression in pituitary adenomas: Role in the regulation of response to somatostatin analogue treatment in patients with acromegaly. Endocrinology 2013, 154, 4715–4725. [Google Scholar] [CrossRef]
- Melmed, S. Medical progress: Acromegaly. New Engl. J. Med. 2006, 355, 2558–2573. [Google Scholar] [CrossRef]
- Katznelson, L.; Laws, E.R., Jr.; Melmed, S.; Molitch, M.E.; Murad, M.H.; Utz, A.; Wass, J.A.; Endocrine, S. Acromegaly: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 3933–3951. [Google Scholar] [CrossRef]
- Jane, J.A., Jr.; Starke, R.M.; Elzoghby, M.A.; Reames, D.L.; Payne, S.C.; Thorner, M.O.; Marshall, J.C.; Laws, E.R., Jr.; Vance, M.L. Endoscopic transsphenoidal surgery for acromegaly: Remission using modern criteria, complications, and predictors of outcome. J. Clin. Endocrinol. Metab. 2011, 96, 2732–2740. [Google Scholar] [CrossRef]
- Nomikos, P.; Buchfelder, M.; Fahlbusch, R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur. J. Endocrinol. 2005, 152, 379–387. [Google Scholar] [CrossRef]
- Giustina, A.; Chanson, P.; Kleinberg, D.; Bronstein, M.D.; Clemmons, D.R.; Klibanski, A.; van der Lely, A.J.; Strasburger, C.J.; Lamberts, S.W.; Ho, K.K.; et al. Expert consensus document: A consensus on the medical treatment of acromegaly. Nat. Rev. Endocrinol. 2014, 10, 243–248. [Google Scholar] [CrossRef]
- Colao, A.; Auriemma, R.S.; Pivonello, R.; Kasuki, L.; Gadelha, M.R. Interpreting biochemical control response rates with first-generation somatostatin analogues in acromegaly. Pituitary 2016, 19, 235–247. [Google Scholar] [CrossRef]
- Murray, R.D.; Kim, K.; Ren, S.G.; Lewis, I.; Weckbecker, G.; Bruns, C.; Melmed, S. The novel somatostatin ligand (SOM230) regulates human and rat anterior pituitary hormone secretion. J. Clin. Endocrinol. Metab. 2004, 89, 3027–3032. [Google Scholar] [CrossRef]
- Hofland, L.J.; van der Hoek, J.; van Koetsveld, P.M.; de Herder, W.W.; Waaijers, M.; Sprij-Mooij, D.; Bruns, C.; Weckbecker, G.; Feelders, R.; van der Lely, A.J.; et al. The novel somatostatin analog SOM230 is a potent inhibitor of hormone release by growth hormone- and prolactin-secreting pituitary adenomas in vitro. J. Clin. Endocrinol. Metab. 2004, 89, 1577–1585. [Google Scholar] [CrossRef]
- Ibanez-Costa, A.; Rivero-Cortes, E.; Vazquez-Borrego, M.C.; Gahete, M.D.; Jimenez-Reina, L.; Venegas-Moreno, E.; de la Riva, A.; Arraez, M.A.; Gonzalez-Molero, I.; Schmid, H.A.; et al. Octreotide and pasireotide (dis)similarly inhibit pituitary tumor cells in vitro. J. Endocrinol. 2016, 231, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Gatto, F.; Feelders, R.A.; Franck, S.E.; van Koetsveld, P.M.; Dogan, F.; Kros, J.M.; Neggers, S.; van der Lely, A.J.; Lamberts, S.W.J.; Ferone, D.; et al. In Vitro Head-to-Head Comparison between Octreotide and Pasireotide in GH-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2017, 102, 2009–2018. [Google Scholar] [CrossRef]
- Colao, A.; Bronstein, M.D.; Freda, P.; Gu, F.; Shen, C.C.; Gadelha, M.; Fleseriu, M.; van der Lely, A.J.; Farrall, A.J.; Hermosillo Resendiz, K.; et al. Pasireotide versus octreotide in acromegaly: A head-to-head superiority study. J. Clin. Endocrinol. Metab. 2014, 99, 791–799. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Bronstein, M.D.; Brue, T.; Coculescu, M.; Fleseriu, M.; Guitelman, M.; Pronin, V.; Raverot, G.; Shimon, I.; Lievre, K.K.; et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): A randomised, phase 3 trial. Lancet. Diabetes Endocrinol. 2014, 2, 875–884. [Google Scholar] [CrossRef]
- Cuny, T.; Barlier, A.; Feelders, R.; Weryha, G.; Hofland, L.J.; Ferone, D.; Gatto, F. Medical therapies in pituitary adenomas: Current rationale for the use and future perspectives. Ann. D’endocrinologie 2015, 76, 43–58. [Google Scholar] [CrossRef]
- Ferone, D.; Gatto, F.; Arvigo, M.; Resmini, E.; Boschetti, M.; Teti, C.; Esposito, D.; Minuto, F. The clinical-molecular interface of somatostatin, dopamine and their receptors in pituitary pathophysiology. J. Mol. Endocrinol. 2009, 42, 361–370. [Google Scholar] [CrossRef]
- Stalla, G.K.; Brockmeier, S.J.; Renner, U.; Newton, C.; Buchfelder, M.; Stalla, J.; Muller, O.A. Octreotide exerts different effects in vivo and in vitro in Cushing’s disease. Eur. J. Endocrinol. 1994, 130, 125–131. [Google Scholar] [CrossRef]
- van der Pas, R.; Feelders, R.A.; Gatto, F.; de Bruin, C.; Pereira, A.M.; van Koetsveld, P.M.; Sprij-Mooij, D.M.; Waaijers, A.M.; Dogan, F.; Schulz, S.; et al. Preoperative normalization of cortisol levels in Cushing’s disease after medical treatment: Consequences for somatostatin and dopamine receptor subtype expression and in vitro response to somatostatin analogs and dopamine agonists. J. Clin. Endocrinol. Metab. 2013, 98, E1880–E1890. [Google Scholar] [CrossRef]
- Gatto, F.; Hofland, L.J. The role of somatostatin and dopamine D2 receptors in endocrine tumors. Endocr. Relat. Cancer 2011, 18, R233–R251. [Google Scholar] [CrossRef] [Green Version]
- van der Hoek, J.; Waaijers, M.; van Koetsveld, P.M.; Sprij-Mooij, D.; Feelders, R.A.; Schmid, H.A.; Schoeffter, P.; Hoyer, D.; Cervia, D.; Taylor, J.E.; et al. Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E278–E287. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, C.; Feelders, R.A.; Waaijers, A.M.; van Koetsveld, P.M.; Sprij-Mooij, D.M.; Lamberts, S.W.; Hofland, L.J. Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J. Mol. Endocrinol. 2009, 42, 47–56. [Google Scholar] [CrossRef]
- Ben-Shlomo, A.; Schmid, H.; Wawrowsky, K.; Pichurin, O.; Hubina, E.; Chesnokova, V.; Liu, N.A.; Culler, M.; Melmed, S. Differential ligand-mediated pituitary somatostatin receptor subtype signaling: Implications for corticotroph tumor therapy. J. Clin. Endocrinol. Metab. 2009, 94, 4342–4350. [Google Scholar] [CrossRef]
- Lamberts, S.W.; de Lange, S.A.; Stefanko, S.Z. Adrenocorticotropin-secreting pituitary adenomas originate from the anterior or the intermediate lobe in Cushing’s disease: Differences in the regulation of hormone secretion. J. Clin. Endocrinol. Metab. 1982, 54, 286–291. [Google Scholar] [CrossRef]
- Pivonello, R.; De Leo, M.; Cozzolino, A.; Colao, A. The Treatment of Cushing’s Disease. Endocr. Rev. 2015, 36, 385–486. [Google Scholar] [CrossRef]
- Boscaro, M.; Ludlam, W.H.; Atkinson, B.; Glusman, J.E.; Petersenn, S.; Reincke, M.; Snyder, P.; Tabarin, A.; Biller, B.M.; Findling, J.; et al. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): A multicenter, phase II trial. J. Clin. Endocrinol. Metab. 2009, 94, 115–122. [Google Scholar] [CrossRef]
- Lacroix, A.; Gu, F.; Gallardo, W.; Pivonello, R.; Yu, Y.; Witek, P.; Boscaro, M.; Salvatori, R.; Yamada, M.; Tauchmanova, L.; et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: A 12 month clinical trial. Lancet. Diabetes Endocrinol. 2018, 6, 17–26. [Google Scholar] [CrossRef]
- Pivonello, R.; Arnaldi, G.; Scaroni, C.; Giordano, C.; Cannavo, S.; Iacuaniello, D.; Trementino, L.; Zilio, M.; Guarnotta, V.; Albani, A.; et al. The medical treatment with pasireotide in Cushing’s disease: An Italian multicentre experience based on “real-world evidence”. Endocrine 2019, 64, 657–672. [Google Scholar] [CrossRef]
- Gatto, F.; Barbieri, F.; Gatti, M.; Wurth, R.; Schulz, S.; Ravetti, J.L.; Zona, G.; Culler, M.D.; Saveanu, A.; Giusti, M.; et al. Balance between somatostatin and D2 receptor expression drives TSH-secreting adenoma response to somatostatin analogues and dopastatins. Clin. Endocrinol. 2012, 76, 407–414. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing but NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef]
- Rindi, G.; Klersy, C.; Albarello, L.; Baudin, E.; Bianchi, A.; Buchler, M.W.; Caplin, M.; Couvelard, A.; Cros, J.; de Herder, W.W.; et al. Competitive Testing of the WHO 2010 versus the WHO 2017 Grading of Pancreatic Neuroendocrine Neoplasms: Data from a Large International Cohort Study. Neuroendocrinology 2018, 107, 375–386. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Kloppel, G. Neuroendocrine Neoplasms: Dichotomy, Origin and Classifications. Visc. Med. 2017, 33, 324–330. [Google Scholar] [CrossRef]
- Oberg, K.; Couvelard, A.; Delle Fave, G.; Gross, D.; Grossman, A.; Jensen, R.T.; Pape, U.F.; Perren, A.; Rindi, G.; Ruszniewski, P.; et al. Antibes Consensus Conference, p., ENETS Consensus Guidelines for Standard of Care in Neuroendocrine Tumours: Biochemical Markers. Neuroendocrinology 2017, 105, 201–211. [Google Scholar] [CrossRef]
- Oberg, K. Neuroendocrine tumors: Recent progress in diagnosis and treatment. Endocr. Relat. Cancer 2011, 18, E3. [Google Scholar] [CrossRef]
- Kloppel, G.; Rindi, G.; Perren, A.; Komminoth, P.; Klimstra, D.S. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: A statement. Virchows Arch. Int. J. Pathol. 2010, 456, 595–597. [Google Scholar] [CrossRef]
- Mai, R.; Kaemmerer, D.; Trager, T.; Neubauer, E.; Sanger, J.; Baum, R.P.; Schulz, S.; Lupp, A. Different somatostatin and CXCR4 chemokine receptor expression in gastroenteropancreatic neuroendocrine neoplasms depending on their origin. Sci. Rep. 2019, 9, 4339. [Google Scholar] [CrossRef]
- Mizutani, G.; Nakanishi, Y.; Watanabe, N.; Honma, T.; Obana, Y.; Seki, T.; Ohni, S.; Nemoto, N. Expression of Somatostatin Receptor (SSTR) Subtypes (SSTR-1, 2A, 3, 4 and 5) in Neuroendocrine Tumors Using Real-time RT-PCR Method and Immunohistochemistry. Acta Histochem. Et Cytochem. 2012, 45, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.R.; Li, T.; Ter-Minassian, M.; Yang, J.; Chan, J.A.; Brais, L.K.; Masugi, Y.; Thiaglingam, A.; Brooks, N.; Nishihara, R.; et al. Association Between Somatostatin Receptor Expression and Clinical Outcomes in Neuroendocrine Tumors. Pancreas 2016, 45, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Sampedro-Nunez, M.; Luque, R.M.; Ramos-Levi, A.M.; Gahete, M.D.; Serrano-Somavilla, A.; Villa-Osaba, A.; Adrados, M.; Ibanez-Costa, A.; Martin-Perez, E.; Culler, M.D.; et al. Presence of sst5TMD4, a truncated splice variant of the somatostatin receptor subtype 5, is associated to features of increased aggressiveness in pancreatic neuroendocrine tumors. Oncotarget 2016, 7, 6593–6608. [Google Scholar] [CrossRef]
- Kaemmerer, D.; Trager, T.; Hoffmeister, M.; Sipos, B.; Hommann, M.; Sanger, J.; Schulz, S.; Lupp, A. Inverse expression of somatostatin and CXCR4 chemokine receptors in gastroenteropancreatic neuroendocrine neoplasms of different malignancy. Oncotarget 2015, 6, 27566–27579. [Google Scholar] [CrossRef]
- Barbieri, F.; Thellung, S.; Wurth, R.; Gatto, F.; Corsaro, A.; Villa, V.; Nizzari, M.; Albertelli, M.; Ferone, D.; Florio, T. Emerging Targets in Pituitary Adenomas: Role of the CXCL12/CXCR4-R7 System. Int. J. Endocrinol. 2014, 2014, 753524. [Google Scholar] [CrossRef]
- Rostene, W.; Guyon, A.; Kular, L.; Godefroy, D.; Barbieri, F.; Bajetto, A.; Banisadr, G.; Callewaere, C.; Conductier, G.; Rovere, C.; et al. Chemokines and chemokine receptors: New actors in neuroendocrine regulations. Front. Neuroendocrinol. 2011, 32, 10–24. [Google Scholar] [CrossRef]
- Werner, R.A.; Weich, A.; Higuchi, T.; Schmid, J.S.; Schirbel, A.; Lassmann, M.; Wild, V.; Rudelius, M.; Kudlich, T.; Herrmann, K.; et al. Imaging of Chemokine Receptor 4 Expression in Neuroendocrine Tumors -a Triple Tracer Comparative Approach. Theranostics 2017, 7, 1489–1498. [Google Scholar] [CrossRef]
- Yao, J.; Wang, J.Y.; Liu, Y.; Wang, B.; Li, Y.X.; Zhang, R.; Wang, L.S.; Liu, L. A randomized phase II study of everolimus for advanced pancreatic neuroendocrine tumors in Chinese patients. Med. Oncol. 2014, 31, 251. [Google Scholar] [CrossRef]
- Faivre, S.; Niccoli, P.; Castellano, D.; Valle, J.W.; Hammel, P.; Raoul, J.L.; Vinik, A.; Van Cutsem, E.; Bang, Y.J.; Lee, S.H.; et al. Sunitinib in pancreatic neuroendocrine tumors: Updated progression-free survival and final overall survival from a phase III randomized study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 339–343. [Google Scholar] [CrossRef]
- Vandamme, T.; Peeters, M.; Dogan, F.; Pauwels, P.; Van Assche, E.; Beyens, M.; Mortier, G.; Vandeweyer, G.; de Herder, W.; Van Camp, G.; et al. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J. Mol. Endocrinol. 2015, 54, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsen, E.; Stridsberg, M.; Taylor, J.E.; Culler, M.D.; Oberg, K.; Janson, E.T. Subtype selective interactions of somatostatin and somatostatin analogs with sst1, sst2, and sst5 in BON-1 cells. Med Oncol. 2004, 21, 285–295. [Google Scholar] [CrossRef]
- Evers, B.M.; Ishizuka, J.; Townsend, C.M.; Thompson, J.C. The human carcinoid cell line, BON. A model system for the study of carcinoid tumors. Ann. New York Acad. Sci. 1994, 733, 393–406. [Google Scholar] [CrossRef]
- Moreno, A.; Akcakanat, A.; Munsell, M.F.; Soni, A.; Yao, J.C.; Meric-Bernstam, F. Antitumor activity of rapamycin and octreotide as single agents or in combination in neuroendocrine tumors. Endocr. -Relat. Cancer 2008, 15, 257–266. [Google Scholar] [CrossRef]
- Herrera-Martinez, A.D.; Gahete, M.D.; Pedraza-Arevalo, S.; Sanchez-Sanchez, R.; Ortega-Salas, R.; Serrano-Blanch, R.; Luque, R.M.; Galvez-Moreno, M.A.; Castano, J.P. Clinical and functional implication of the components of somatostatin system in gastroenteropancreatic neuroendocrine tumors. Endocrine 2018, 59, 426–437. [Google Scholar] [CrossRef]
- Herrera-Martinez, A.D.; Hofland, J.; Hofland, L.J.; Brabander, T.; Eskens, F.; Galvez Moreno, M.A.; Luque, R.M.; Castano, J.P.; de Herder, W.W.; Feelders, R.A. Targeted Systemic Treatment of Neuroendocrine Tumors: Current Options and Future Perspectives. Drugs 2019, 79, 21–42. [Google Scholar] [CrossRef]
- van Adrichem, R.C.; de Herder, W.W.; Kamp, K.; Brugts, M.P.; de Krijger, R.R.; Sprij-Mooij, D.M.; Lamberts, S.W.; van Koetsveld, P.M.; Janssen, J.A.; Hofland, L.J. Effects of Somatostatin Analogs and Dopamine Agonists on Insulin-Like Growth Factor 2-Induced Insulin Receptor Isoform A Activation by Gastroenteropancreatic Neuroendocrine Tumor Cells. Neuroendocrinology 2016, 103, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Hofving, T.; Arvidsson, Y.; Almobarak, B.; Inge, L.; Pfragner, R.; Persson, M.; Stenman, G.; Kristiansson, E.; Johanson, V.; Nilsson, O. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr. Relat. Cancer 2018, 25, X1–X2. [Google Scholar] [CrossRef] [Green Version]
- Cambiaghi, V.; Vitali, E.; Morone, D.; Peverelli, E.; Spada, A.; Mantovani, G.; Lania, A.G. Identification of human somatostatin receptor 2 domains involved in internalization and signaling in QGP-1 pancreatic neuroendocrine tumor cell line. Endocrine 2017, 56, 146–157. [Google Scholar] [CrossRef]
- Hofland, L.J.; Lamberts, S.W. The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr. Rev. 2003, 24, 28–47. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schonbrunn, A. Illuminating somatostatin analog action at neuroendocrine tumor receptors. Trends Pharm. Sci. 2013, 34, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.; Romano, D.; Saveanu, A.; Roche, C.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Garcia, S.; et al. Anti-proliferative and anti-secretory effects of everolimus on human pancreatic neuroendocrine tumors primary cultures: Is there any benefit from combination with somatostatin analogs? Oncotarget 2017, 8, 41044–41063. [Google Scholar] [CrossRef]
- Wolin, E.M. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013, 335, 1–8. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Grozinsky-Glasberg, S.; Franchi, G.; Teng, M.; Leontiou, C.A.; Ribeiro de Oliveira, A., Jr.; Dalino, P.; Salahuddin, N.; Korbonits, M.; Grossman, A.B. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell Line. Neuroendocrinology 2008, 87, 168–181. [Google Scholar] [CrossRef]
- Lewis, C.S.; Elnakat Thomas, H.; Orr-Asman, M.A.; Green, L.C.; Boody, R.E.; Matiash, K.; Karve, A.; Hisada, Y.M.; Davis, H.W.; Qi, X.; et al. mTOR kinase inhibition reduces tissue factor expression and growth of pancreatic neuroendocrine tumors. J. Thromb. Haemost. JTH 2019, 17, 169–182. [Google Scholar] [CrossRef]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef]
- Lee, L.; Ito, T.; Jensen, R.T. Everolimus in the treatment of neuroendocrine tumors: Efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin. Pharmacother. 2018, 19, 909–928. [Google Scholar] [CrossRef]
- Perren, A.; Couvelard, A.; Scoazec, J.Y.; Costa, F.; Borbath, I.; Delle Fave, G.; Gorbounova, V.; Gross, D.; Grossma, A.; Jense, R.T.; et al. Antibes Consensus Conference, p. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Pathology: Diagnosis and Prognostic Stratification. Neuroendocrinology 2017, 105, 196–200. [Google Scholar] [CrossRef]
- Pavel, M.; Valle, J.W.; Eriksson, B.; Rinke, A.; Caplin, M.; Chen, J.; Costa, F.; Falkerby, J.; Fazio, N.; Gorbounova, V.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms: Systemic Therapy-Biotherapy and Novel Targeted Agents. Neuroendocrinology 2017, 105, 266–280. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Rinke, A.; Valle, J.W.; Fazio, N.; Caplin, M.; Gorbounova, V.; J, O.C.; Eriksson, B.; Sorbye, H.; Kulke, M.; et al. Antibes Consensus Conference, p. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms. Systemic Therapy 2: Chemotherapy. Neuroendocrinology 2017, 105, 281–294. [Google Scholar] [CrossRef]
- Zandee, W.T.; Brabander, T.; Blazevic, A.; Kam, B.L.R.; Teunissen, J.J.M.; Feelders, R.A.; Hofland, J.; de Herder, W.W. Symptomatic and Radiological Response to 177Lu-DOTATATE for the Treatment of Functioning Pancreatic Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1336–1344. [Google Scholar] [CrossRef]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.; Toumpanakis, C.; Kaltsas, G.; et al. Antibes Consensus Conference, p., ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasia: Peptide Receptor Radionuclide Therapy with Radiolabeled Somatostatin Analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef]
- Thang, S.P.; Lung, M.S.; Kong, G.; Hofman, M.S.; Callahan, J.; Michael, M.; Hicks, R.J. Peptide receptor radionuclide therapy (PRRT) in European Neuroendocrine Tumour Society (ENETS) grade 3 (G3) neuroendocrine neoplasia (NEN)-a single-institution retrospective analysis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 262–277. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Halfdanarson, T.R.; Bellizzi, A.M.; Chan, J.A.; Dillon, J.S.; Heaney, A.P.; Kunz, P.L.; O’Dorisio, T.M.; Salem, R.; Segelov, E.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Medical Management of Midgut Neuroendocrine Tumors. Pancreas 2017, 46, 707–714. [Google Scholar] [CrossRef]
- Hofland, J.; Herrera Martinez, A.D.; Zandee, W.T.; de Herder, W.W. Management of carcinoid syndrome: A systematic review and meta-analysis. Endocr. Relat. Cancer 2019. [Google Scholar] [CrossRef]
- Khan, M.S.; El-Khouly, F.; Davies, P.; Toumpanakis, C.; Caplin, M.E. Long-term results of treatment of malignant carcinoid syndrome with prolonged release Lanreotide (Somatuline Autogel). Aliment. Pharmacol. Ther. 2011, 34, 235–242. [Google Scholar] [CrossRef]
- Ruszniewski, P.; O’Toole, D. Ablative therapies for liver metastases of gastroenteropancreatic endocrine tumors. Neuroendocrinology 2004, 80, 74–78. [Google Scholar] [CrossRef]
- Ruszniewski, P.; Valle, J.W.; Lombard-Bohas, C.; Cuthbertson, D.J.; Perros, P.; Holubec, L.; Delle Fave, G.; Smith, D.; Niccoli, P.; Maisonobe, P.; et al. Patient-reported outcomes with lanreotide Autogel/Depot for carcinoid syndrome: An international observational study. Dig. Liver Dis. 2016, 48, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Godara, A.; Siddiqui, N.S.; Byrne, M.M.; Saif, M.W. The safety of lanreotide for neuroendocrine tumor. Expert Opin. Drug Saf. 2019, 18, 1–10. [Google Scholar] [CrossRef]
- Modlin, I.M.; Pavel, M.; Kidd, M.; Gustafsson, B.I. Review article: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment. Pharm. Ther. 2010, 31, 169–188. [Google Scholar]
- Li, S.C.; Martijn, C.; Cui, T.; Essaghir, A.; Luque, R.M.; Demoulin, J.B.; Castano, J.P.; Oberg, K.; Giandomenico, V. The somatostatin analogue octreotide inhibits growth of small intestine neuroendocrine tumour cells. PLoS ONE 2012, 7, e48411. [Google Scholar] [CrossRef]
- Chan, D.L.; Segelov, E.; Singh, S. Everolimus in the management of metastatic neuroendocrine tumours. Ther. Adv. Gastroenterol. 2017, 10, 132–141. [Google Scholar] [CrossRef]
- Kvols, L.K.; Oberg, K.E.; O’Dorisio, T.M.; Mohideen, P.; de Herder, W.W.; Arnold, R.; Hu, K.; Zhang, Y.; Hughes, G.; Anthony, L.; et al. Pasireotide (SOM230) shows efficacy and tolerability in the treatment of patients with advanced neuroendocrine tumors refractory or resistant to octreotide LAR: Results from a phase II study. Endocr. Relat. Cancer 2012, 19, 657–666. [Google Scholar] [CrossRef]
- Wolin, E.M.; Jarzab, B.; Eriksson, B.; Walter, T.; Toumpanakis, C.; Morse, M.A.; Tomassetti, P.; Weber, M.M.; Fogelman, D.R.; Ramage, J.; et al. Phase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analogues. Drug Des. Dev. Ther. 2015, 9, 5075–5086. [Google Scholar] [CrossRef] [Green Version]
- Kulke, M.H.; Ruszniewski, P.; Van Cutsem, E.; Lombard-Bohas, C.; Valle, J.W.; De Herder, W.W.; Pavel, M.; Degtyarev, E.; Brase, J.C.; Bubuteishvili-Pacaud, L.; et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-differentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann. Oncol. 2017, 28, 1309–1315. [Google Scholar] [CrossRef]
- Yao, J.C.; Chan, J.A.; Mita, A.C.; Kundu, M.G.; Hermosillo Resendiz, K.; Hu, K.; Ravichandran, S.; Strosberg, J.R.; Wolin, E.M. Phase I dose-escalation study of long-acting pasireotide in patients with neuroendocrine tumors. OncoTargets Ther. 2017, 10, 3177–3186. [Google Scholar] [CrossRef]
- Cives, M.; Kunz, P.L.; Morse, B.; Coppola, D.; Schell, M.J.; Campos, T.; Nguyen, P.T.; Nandoskar, P.; Khandelwal, V.; Strosberg, J.R. Phase II clinical trial of pasireotide long-acting repeatable in patients with metastatic neuroendocrine tumors. Endocr. Relat. Cancer 2015, 22, 1–9. [Google Scholar] [CrossRef]
- Ferolla, P.; Brizzi, M.P.; Meyer, T.; Mansoor, W.; Mazieres, J.; Do Cao, C.; Lena, H.; Berruti, A.; Damiano, V.; Buikhuisen, W.; et al. Efficacy and safety of long-acting pasireotide or everolimus alone or in combination in patients with advanced carcinoids of the lung and thymus (LUNA): An open-label, multicentre, randomised, phase 2 trial. Lancet. Oncol. 2017, 18, 1652–1664. [Google Scholar] [CrossRef]
- Kaderli, R.M.; Spanjol, M.; Kollar, A.; Butikofer, L.; Gloy, V.; Dumont, R.A.; Seiler, C.A.; Christ, E.R.; Radojewski, P.; Briel, M.; et al. Therapeutic Options for Neuroendocrine Tumors: A Systematic Review and Network Meta-analysis. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Wurth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef]
- Wurth, R.; Barbieri, F.; Pattarozzi, A.; Gaudenzi, G.; Gatto, F.; Fiaschi, P.; Ravetti, J.L.; Zona, G.; Daga, A.; Persani, L.; et al. Phenotypical and Pharmacological Characterization of Stem-Like Cells in Human Pituitary Adenomas. Mol. Neurobiol. 2017, 54, 4879–4895. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SST2 Trafficking Properties | Ligand | Receptor Internalization (EC50, nM) | Maximal Internalization (% of total) a | Receptor Recycling (min) b |
|---|---|---|---|---|
| hSST2 * | SRIF | 3.26 | − | n.a. |
| OCT | 6.46 | − | n.a. | |
| PAS | 31.78 | − | n.a. | |
| rSST2 ** | SRIF | 0.4 | 82.8 | 42.3 |
| OCT | n.a. | n.a. | n.a. | |
| PAS | 23.3 | 46.0 | 4.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatto, F.; Barbieri, F.; Arvigo, M.; Thellung, S.; Amarù, J.; Albertelli, M.; Ferone, D.; Florio, T. Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. Int. J. Mol. Sci. 2019, 20, 3940. https://doi.org/10.3390/ijms20163940
Gatto F, Barbieri F, Arvigo M, Thellung S, Amarù J, Albertelli M, Ferone D, Florio T. Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. International Journal of Molecular Sciences. 2019; 20(16):3940. https://doi.org/10.3390/ijms20163940
Chicago/Turabian StyleGatto, Federico, Federica Barbieri, Marica Arvigo, Stefano Thellung, Jessica Amarù, Manuela Albertelli, Diego Ferone, and Tullio Florio. 2019. "Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms" International Journal of Molecular Sciences 20, no. 16: 3940. https://doi.org/10.3390/ijms20163940
APA StyleGatto, F., Barbieri, F., Arvigo, M., Thellung, S., Amarù, J., Albertelli, M., Ferone, D., & Florio, T. (2019). Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. International Journal of Molecular Sciences, 20(16), 3940. https://doi.org/10.3390/ijms20163940