Synthesis of New Quinoline-Piperonal Hybrids as Potential Drugs against Alzheimer’s Disease

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
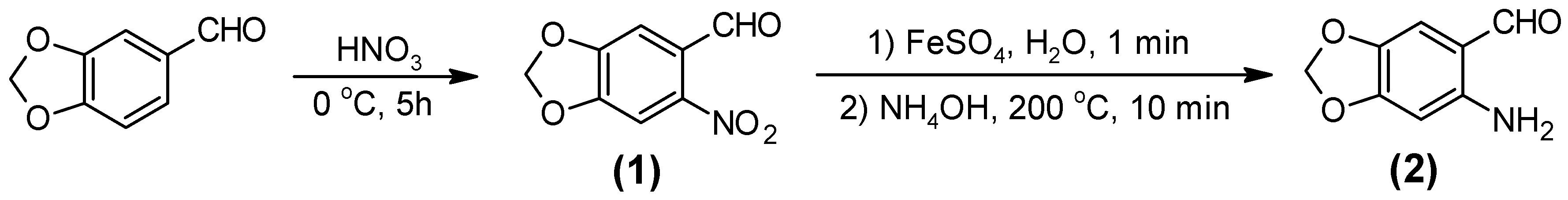
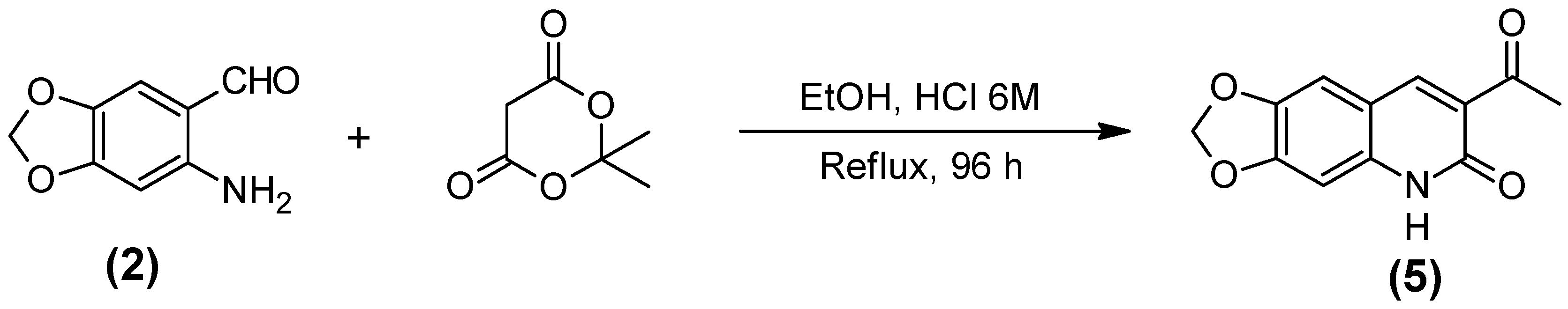
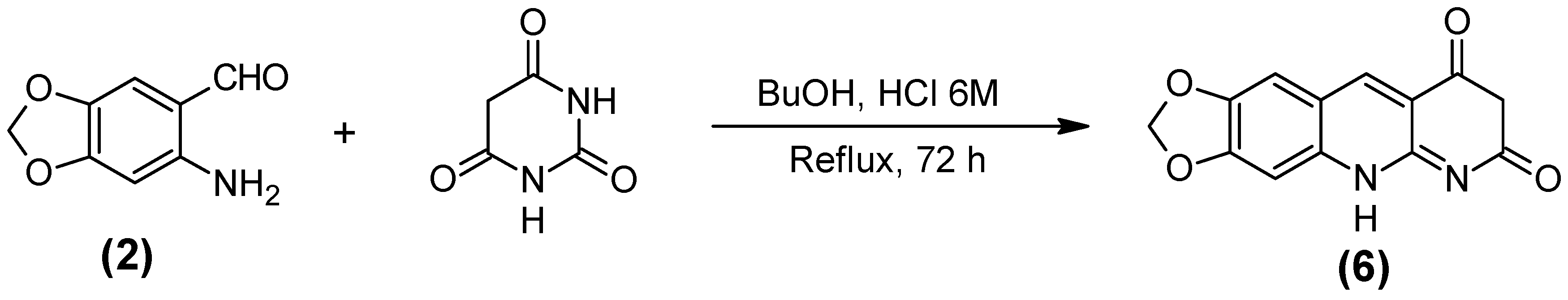
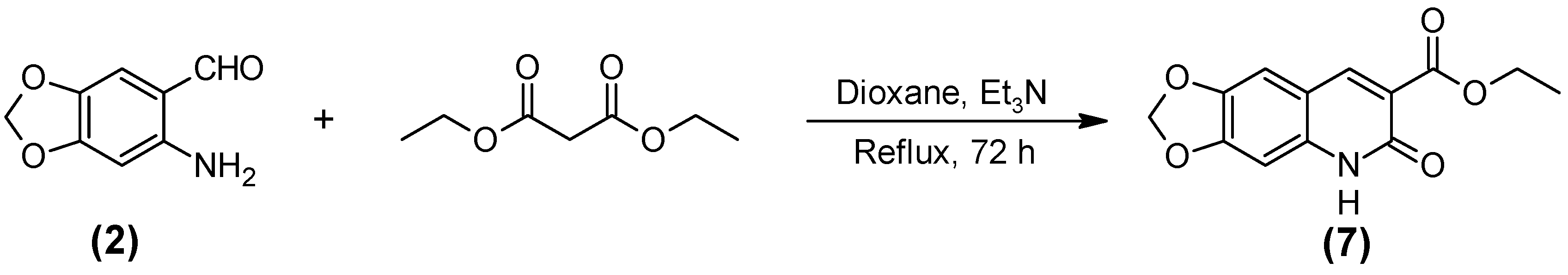
2.1. Synthesis of the Quinoline-Piperonal Hybrids
2.2. Calculation of Pharmacokinetic and Toxicological Properties
2.3. Cholinesterase Inhibition Assay through Ellman’s Method
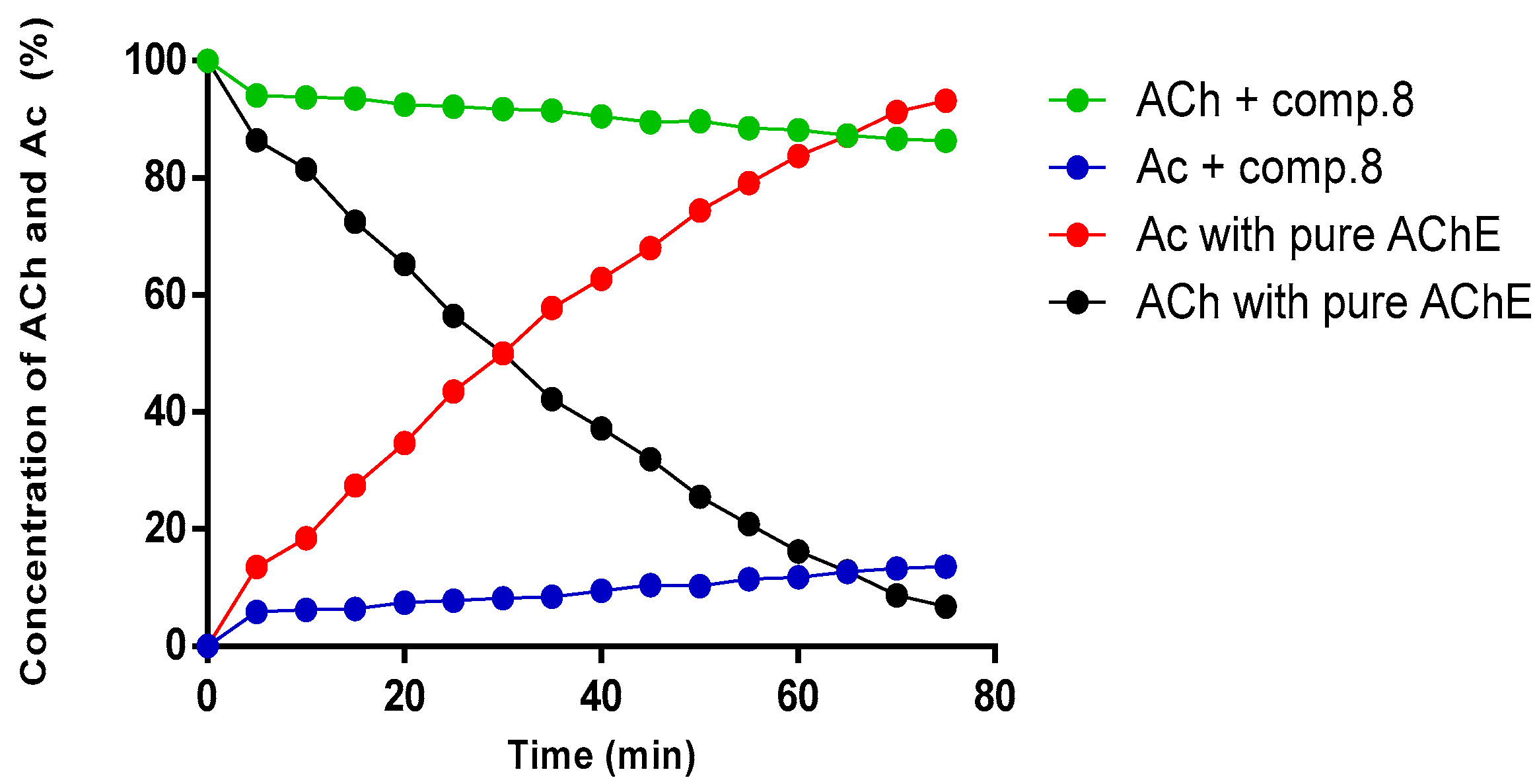
2.4. Kinetic Study of Compound (8) by NMR
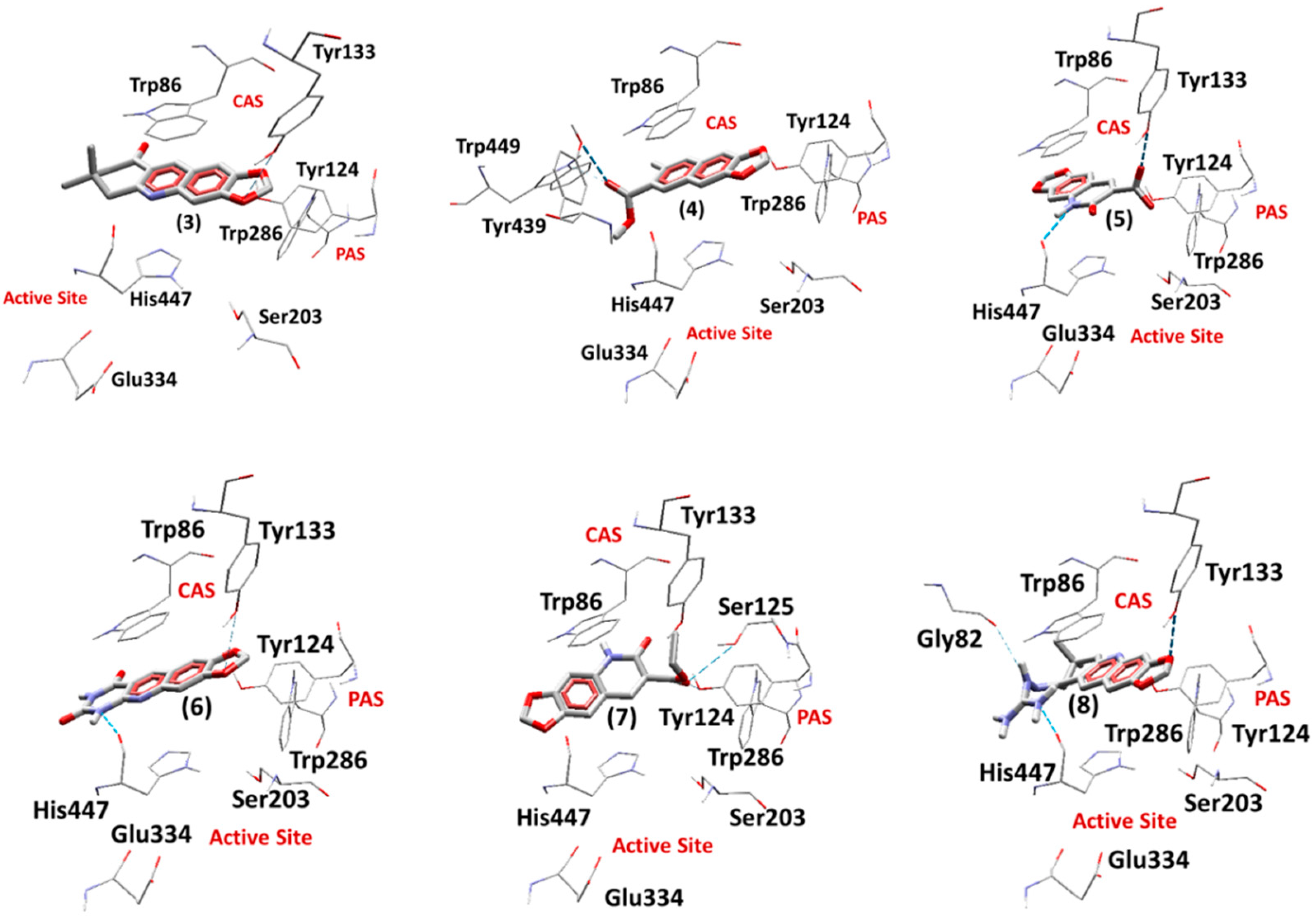
2.5. Docking Studies
2.5.1. Docking Studies on EeAChE
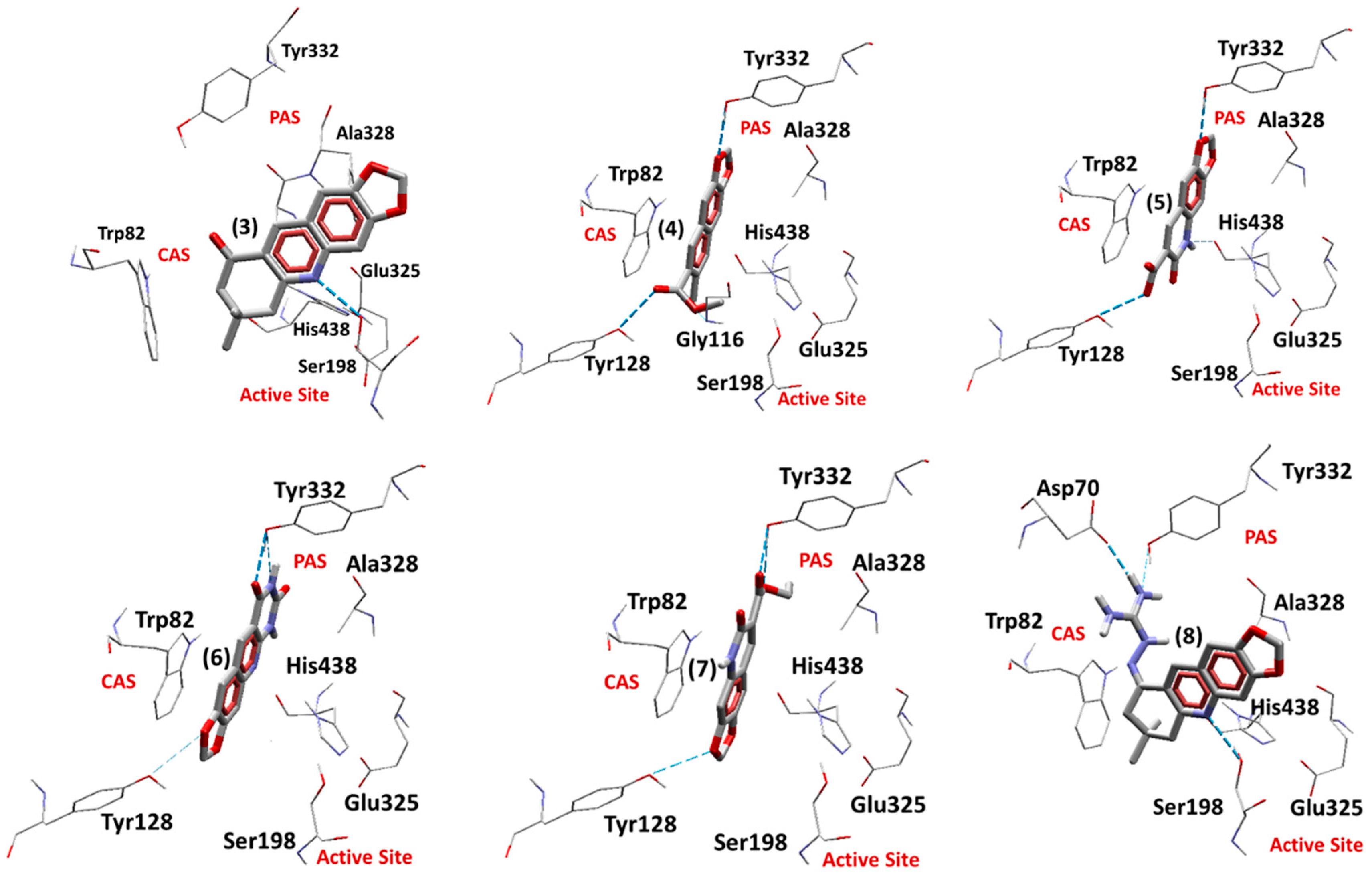
2.5.2. Docking Studies on EqBChE
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of The Compounds
3.2.1. 6-Nitro-1,3-benzodioxole-5-carboxaldehide [6-nitropiperonal, (1)]
3.2.2. 6-Amino-1,3-benzodioxole-5-carboxaldehide [(6-aminopiperonal, (2)]
3.2.3. 7,8-Dihydro-7,7-dimethyl-1,3-dioxolo[4,5-b]acridin-9(6h)-one (3)
3.2.4. Methyl 6-Methyl-[1,3]dioxolo[4,5-g]quinone-7-carboxilate (4)
3.2.5. Acid 5,6-Diidro-6-oxo-1,3-dioxolo[4,6g] quiniline-7-carboxilic (5)
3.2.6. 1,3-Dioxolo [4,5-g] pirimido [4,5-b] quinoline-7,9 (6H,8H)-dione (6)
3.2.7. Ethyl 5,6-Dehidro-6-oxo-[1,3] dioxolano [4,5-g] quinoline-7-carboxilate (7)
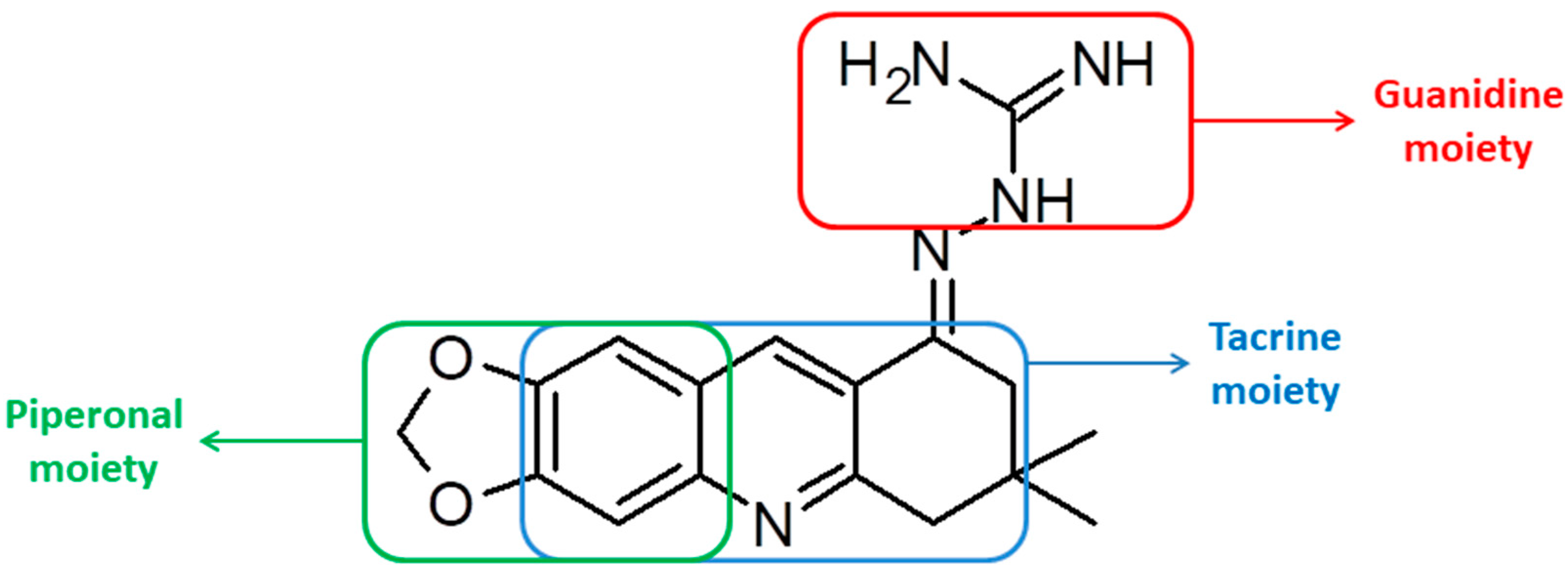
3.2.8. Guanylhydrazone of 7,7-Dimethyl-7,8-dihydro-[1,3]dioxolo[4,5-b]acridin-9(6h)-one (8)
3.3. NMR Analysis
3.4. Structure Optimization and Calculation of Pharmacokinetic and Toxicological Properties
3.5. Cholinesterase Inhibition Assay by Ellman’s Method
3.6. AChE Inhibition Assay by NMR Method
3.7. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| AD | Alzheimer Disease |
| ATCI | Acetylthiocholine Iodide |
| BBB | Blood Brain Barrier |
| BChE | Butyrilcholinesterase |
| BTCI | Butyrylthiocholine Iodide |
| CAS | Catalytic Anionic Site |
| CNS | Central Nervous System |
| DFT | Density Functional Theory |
| DMSO | Dimethylsulfoxide |
| DMSO-d6 | Deuterated DMSO |
| DTNB | 5,5-dithio-bis-(2-nitrobenzoic acid) |
| EeAChE | Electrophorus electricus AChE |
| EqBChE | Equine serum BChE |
| IR | Infrared |
| MHz | Megahertz |
| NMR | Nuclear Magnetic Ressonance |
| PAS | Peripheral Anionic Site |
| PDB | Protein Data Bank |
| PSA | Polar Surface Area |
| RMSD | Root Mean Square Deviation |
| TLC | Thin Layer Chromatography |
References
- Burns, A.; Byrne, E.J.; Maurer, K. Alzheimer’s disease. Lancet 2002, 360, 163–165. [Google Scholar] [CrossRef]
- Kuca, K.; Soukup, O.; Maresova, P.; Korabecny, J.; Nepovimova, E.; Klimova, B.; Honegr, J.; Ramalho, T.C.; França, T.C.C. Current Approaches Against Alzheimer’s Disease in Clinical Trials. J. Braz. Chem. Soc. 2016, 27, 641–649. [Google Scholar] [CrossRef]
- Maresova, P.; Mohelska, H.; Dolejs, J.; Kuca, K. Socio-economic Aspects of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Dalvi, A. Alzheimer’s Disease. Disease-a-Month 2012, 58, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Curr. Ther. Res. 2003, 64, 216–235. [Google Scholar] [CrossRef]
- Cheffer, A.; Ulrich, H. Inhibition Mechanism of Rat α3β4 Nicotinic Acetylcholine Receptor by the Alzheimer Therapeutic Tacrine. Biochemistry 2011, 50, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Horova, A.; Musilek, K.; Nepovimova, E.; Drtinova, L.; Gazova, Z.; Siposova, K.; Dolezal, R.; Jun, D.; et al. Design, synthesis and in vitro testing of 7-methoxytacrine-amantadine analogues: a novel cholinesterase inhibitors for the treatment of Alzheimer’s disease. Med. Chem. Res. 2015, 24, 2645–2655. [Google Scholar] [CrossRef]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Nguyen, T.D.; Jun, D.; Soukup, O.; Pasdiorova, M.; Jost, P.; Muckova, L.; Malinak, D.; et al. A 7-methoxytacrine–4-pyridinealdoxime hybrid as a novel prophylactic agent with reactivation properties in organophosphate intoxication. Toxicol. Res. 2016, 5, 1012–1016. [Google Scholar] [CrossRef]
- Kumar, S.; Bawa, S.; Gupta, H. Biological Activities of Quinoline Derivatives. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Fraga, C.A.M.; Miranda, A.L.P.; Rodrigues, C.R. A química medicinal de N-acilidrazonas: Novos compostos-protótipos de fármacos analgésicos, antiinflamatórios e anti-trombóticos. Quim. Nov. 2002, 25, 129–148. [Google Scholar] [CrossRef]
- Petronilho, E.d.C.; Rennó, M.N.; Castro, N.G.; da Silva, F.M.R.; Pinto, A.d.C.; Figueroa-Villar, J.D. Design, synthesis, and evaluation of guanylhydrazones as potential inhibitors or reactivators of acetylcholinesterase. J. Enzyme Inhib. Med. Chem. 2016, 31, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Ekeley, J.B.; Klemme, M.S. The nitration of piperonal. J. Am. Chem. Soc. 1928, 50, 2711–2715. [Google Scholar] [CrossRef]
- Bogert, M.T.; Elder, F.R. The synthesis of 6-hydroxypiperonyli acid and incidental compounds. J. Am. Chem. Soc. 1929, 51, 532–539. [Google Scholar] [CrossRef]
- Jacobs, W.A.; Heidelberger, M. The Ferrous sulfate and ammonia method for the reduction of nitro to amino compounds. J. Am. Chem. Soc. 1917, 39, 1435–1439. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Pérez-Mayoral, E.; Samadi, A.; Carreiras, M.d.C.; Soriano, E. Recent Advances in the Friedländer Reaction. Chem. Rev. 2009, 109, 2652–2671. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Sachan, N.; Chawla, P. Synthesis and Evaluation of Thiazolidinedione-Coumarin Adducts as Antidiabetic, Anti-Inflammatory and Antioxidant Agents. Lett. Org. Chem. 2015, 12, 429–455. [Google Scholar] [CrossRef]
- Corma, A.; Martín-Aranda, R.M. Alkaline-substituted sepiolites as a new type of strong base catalyst. J. Catal. 1991, 130, 130–137. [Google Scholar] [CrossRef]
- Borges, M.N.; Messeder, J.C.; Figueroa-Villar, J.D. Synthesis, anti-Trypanosoma cruzi activity and micelle interaction studies of bisguanylhydrazones analogous to pentamidine. Eur. J. Med. Chem. 2004, 39, 925–929. [Google Scholar] [CrossRef]
- Hehre, W.J.; Deppmeier, B.J. PC SPARTAN Pro. Wavefunction, Inc., Irvine, 1999; Wavefunction: Irvine, CA, USA, 1999. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, C.; Yang, J. Predictive model of blood-brain barrier penetration of organic compounds1. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira Neto, D.C.; de Souza Ferreira, M.; da Conceição Petronilho, E.; Alencar Lima, J.; Oliveira Francisco de Azeredo, S.; de Oliveira Carneiro Brum, J.; Jorge do Nascimento, C.; Figueroa Villar, J.D. A new guanylhydrazone derivative as a potential acetylcholinesterase inhibitor for Alzheimer’s disease: Synthesis, molecular docking, biological evaluation and kinetic studies by nuclear magnetic resonance. RSC Adv. 2017, 7, 33944–33952. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Pacheco, G.; Palacios-Esquivel, R.; Moss, D.E. Cholinesterase inhibitors proposed for treating dementia in Alzheimer’s disease: Selectivity toward human brain acetylcholinesterase compared with butyrylcholinesterase. J. Pharmacol. Exp. Ther. 1995, 274, 767–770. [Google Scholar] [PubMed]
- Pagliosa, L.B.; Monteiro, S.C.; Silva, K.B.; de Andrade, J.P.; Dutilh, J.; Bastida, J.; Cammarota, M.; Zuanazzi, J.A.S. Effect of isoquinoline alkaloids from two Hippeastrum species on in vitro acetylcholinesterase activity. Phytomedicine 2010, 17, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Komloova, M.; Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Structure-Activity Relationship of Quaternary Acetylcholinesterase Inhibitors—Outlook for Early Myasthenia Gravis Treatment. Curr. Med. Chem. 2010, 17, 1810–1824. [Google Scholar] [CrossRef]
- Sugimoto, H.; Tsuchiya, Y.; Sugumi, H.; Higurashi, K.; Karibe, N.; Iimura, Y.; Sasaki, A.; Araki, S.; Yamanishi, Y.; Yamatsu, K. Synthesis and structure-activity relationships of acetylcholinesterase inhibitors: 1-benzyl-4-(2-phthalimidoethyl)piperidine, and related derivatives. J. Med. Chem. 1992, 35, 4542–4548. [Google Scholar] [CrossRef]
- Tumiatti, V.; Rosini, M.; Bartolini, M.; Cavalli, A.; Marucci, G.; Andrisano, V.; Angeli, P.; Banzi, R.; Minarini, A.; Recanatini, M.; et al. Structure−Activity Relationships of Acetylcholinesterase Noncovalent Inhibitors Based on a Polyamine Backbone. 2. Role of the Substituents on the Phenyl Ring and Nitrogen Atoms of Caproctamine. J. Med. Chem. 2003, 46, 954–966. [Google Scholar] [CrossRef]
- Feitosa Da Cunha, S.; Soares, X.; Vieira, A.A.A.; Delfino, R.T.T.; Figueroa-Villar, D. NMR determination of Electrophorus electricus acetylcholinesterase inhibition and reactivation by neutral oximes. Bioorg. Med. Chem. 2013, 21, 5923–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Wagner, G. NMR studies of protein interactions. Curr. Opin. Struct. Biol. 2006, 16, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Goldflam, M.; Tarragó, T.; Gairí, M.; Giralt, E. NMR Studies of Protein–Ligand Interactions. Methods Mol. Biol. 2012, 831, 233–259. [Google Scholar] [PubMed]
- Figueroa-Villar, J.D. Design, synthesis, structure, toxicology and in vitro testing of three novel agents for Alzheimer’s disease. RSC Adv. 2017, 7, 23457–23467. [Google Scholar] [CrossRef]
- Ferreira Neto, D.C.; Alencar Lima, J.; Sobreiro Francisco Diz de Almeida, J.; Costa França, T.C.; Jorge do Nascimento, C.; Figueroa Villar, J.D. New semicarbazones as gorge-spanning ligands of acetylcholinesterase and potential new drugs against Alzheimer’s disease: Synthesis, molecular modeling, NMR, and biological evaluation. J. Biomol. Struct. Dyn. 2018, 36, 4099–4113. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.N.; Hopper, P.F.; Campbell, B.K. The Preparation of Methylenedioxy-Methoxybenzaldehydes. J. Org. Chem. 1951, 16, 1736–1741. [Google Scholar] [CrossRef]
- Lima, J.A.; Costa, R.S.; Epifânio, R.A.; Castro, N.G.; Rocha, M.S.; Pinto, A.C. Geissospermum vellosii stembark. Pharmacol. Biochem. Behav. 2009, 92, 508–513. [Google Scholar] [CrossRef]
- Swain, M. chemicalize.orgc hemicalize.org by ChemAxon Ltd. J. Chem. Inf. Model. 2012, 52, 613–615. [Google Scholar] [CrossRef]
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]













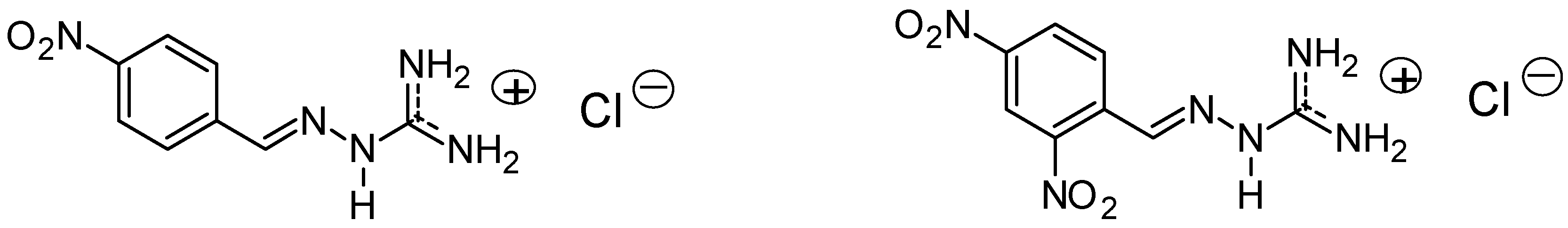{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
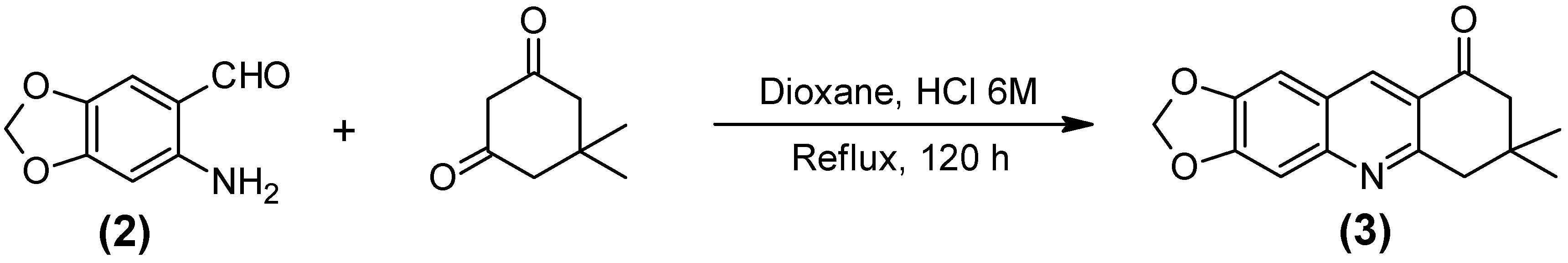{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
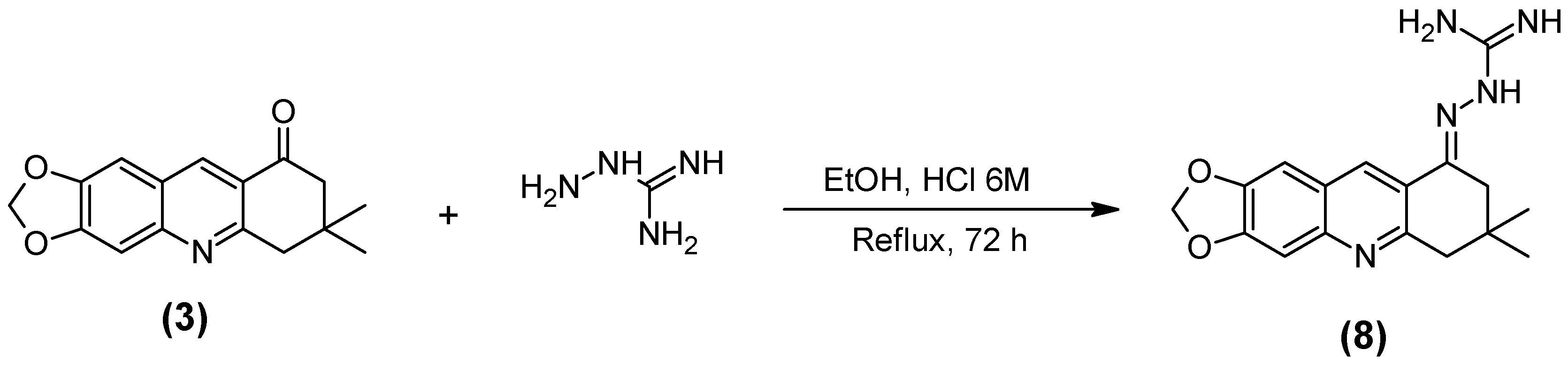{kind=link}
| Comp | Weight (amu) | Area (Å2) | Volume (Å3) | PSA (Å2) | Acute Toxicity (algae) | Carcinog. (mouse) | Meet the Lipinski’s Rule? | BBB Penetration |
|---|---|---|---|---|---|---|---|---|
| 3 | 269.30 | 275.40 | 266.12 | 38.55 | 0.05 | Negative | Yes | 2.69 |
| 4 | 245.23 | 250.08 | 233.59 | 45.63 | 0.09 | Negative | Yes | 1.37 |
| 5 | 233.18 | 216.47 | 202.36 | 70.70 | 0.15 | Positive | Yes | 0.29 |
| 6 | 257.21 | 235.21 | 221.47 | 75.45 | 0.19 | Positive | Yes | 0.39 |
| 7 | 261.23 | 260.62 | 242.00 | 63.40 | 0.12 | Negative | Yes | 0.04 |
| 8 | 325.37 | 334.47 | 317.07 | 89.56 | 0.03 | Negative | Yes | 0.11 |
| Tacrine | 198.27 | 225.26 | 213.94 | 30.24 | 0.05 | Positive | Yes | 0.86 |
| Compound | EeAChE | EqBChE |
|---|---|---|
| (3) | 51.51 ± 2.47 | - |
| (4) | - | - |
| (5) | 14.15 ± 1.15 | - |
| (6) | 32.06 ± 3.53 | - |
| (7) | 62.76 ± 2.02 | - |
| (8) | 8.50 ± 0.39 | 15.16 ± 0.75 |
| Tacrine | 0.0414 ± 0.0002 | 0.0045 ± 0.0005 |
| Ligand | Einter (Kcal.mol−1) | EH-bond (Kcal.mol−1) | H-bond Interactions | RMSD (Å) |
|---|---|---|---|---|
| Redocking Tacrine | −132.39 | −0.20 | His447 | 0.28 |
| (3) | −132.69 | −0.13 | Tyr133 | - |
| (4) | −135.51 | −0.18 | Trp439, Tyr449 | - |
| (5) | −140.88 | −0.22 | Tyr133 | - |
| (6) | −145.33 | −1.76 | Tyr133, His447 | - |
| (7) | −119.49 | −0.46 | Tyr124, Ser125 | - |
| (8) | −155.88 | −2.35 | Tyr133, His447 | - |
| Ligand | Einter (Kcal.mol−1) | EH-bond (Kcal.mol−1) | H-bond Interactions | RMSD (Å) |
|---|---|---|---|---|
| Redocking Tacrine | −133.63 | −2.08 | His438 | 0.30 |
| (3) | −153.23 | −2.50 | Ser198 | - |
| (4) | −152.11 | −5.27 | Gly116, Tyr128, Tyr332 | - |
| (5) | −155.08 | −5.73 | Tyr128, Tyr332, His438 | - |
| (6) | −151.08 | −3.96 | Tyr128, Tyr332 | - |
| (7) | −158.06 | −4.26 | Tyr128, Tyr332 | - |
| (8) | −196.99 | −5.43 | Asp70, Ser198, Tyr332 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira C. Brum, J.; Neto, D.C.F.; de Almeida, J.S.F.D.; Lima, J.A.; Kuca, K.; França, T.C.C.; Figueroa-Villar, J.D. Synthesis of New Quinoline-Piperonal Hybrids as Potential Drugs against Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3944. https://doi.org/10.3390/ijms20163944
de Oliveira C. Brum J, Neto DCF, de Almeida JSFD, Lima JA, Kuca K, França TCC, Figueroa-Villar JD. Synthesis of New Quinoline-Piperonal Hybrids as Potential Drugs against Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(16):3944. https://doi.org/10.3390/ijms20163944
Chicago/Turabian Stylede Oliveira C. Brum, Juliana, Denise Cristian F. Neto, Joyce Sobreiro F. D. de Almeida, Josélia Alencar Lima, Kamil Kuca, Tanos Celmar C. França, and José D. Figueroa-Villar. 2019. "Synthesis of New Quinoline-Piperonal Hybrids as Potential Drugs against Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 16: 3944. https://doi.org/10.3390/ijms20163944
APA Stylede Oliveira C. Brum, J., Neto, D. C. F., de Almeida, J. S. F. D., Lima, J. A., Kuca, K., França, T. C. C., & Figueroa-Villar, J. D. (2019). Synthesis of New Quinoline-Piperonal Hybrids as Potential Drugs against Alzheimer’s Disease. International Journal of Molecular Sciences, 20(16), 3944. https://doi.org/10.3390/ijms20163944