Matrisome Properties of Scaffolds Direct Fibroblasts in Idiopathic Pulmonary Fibrosis

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
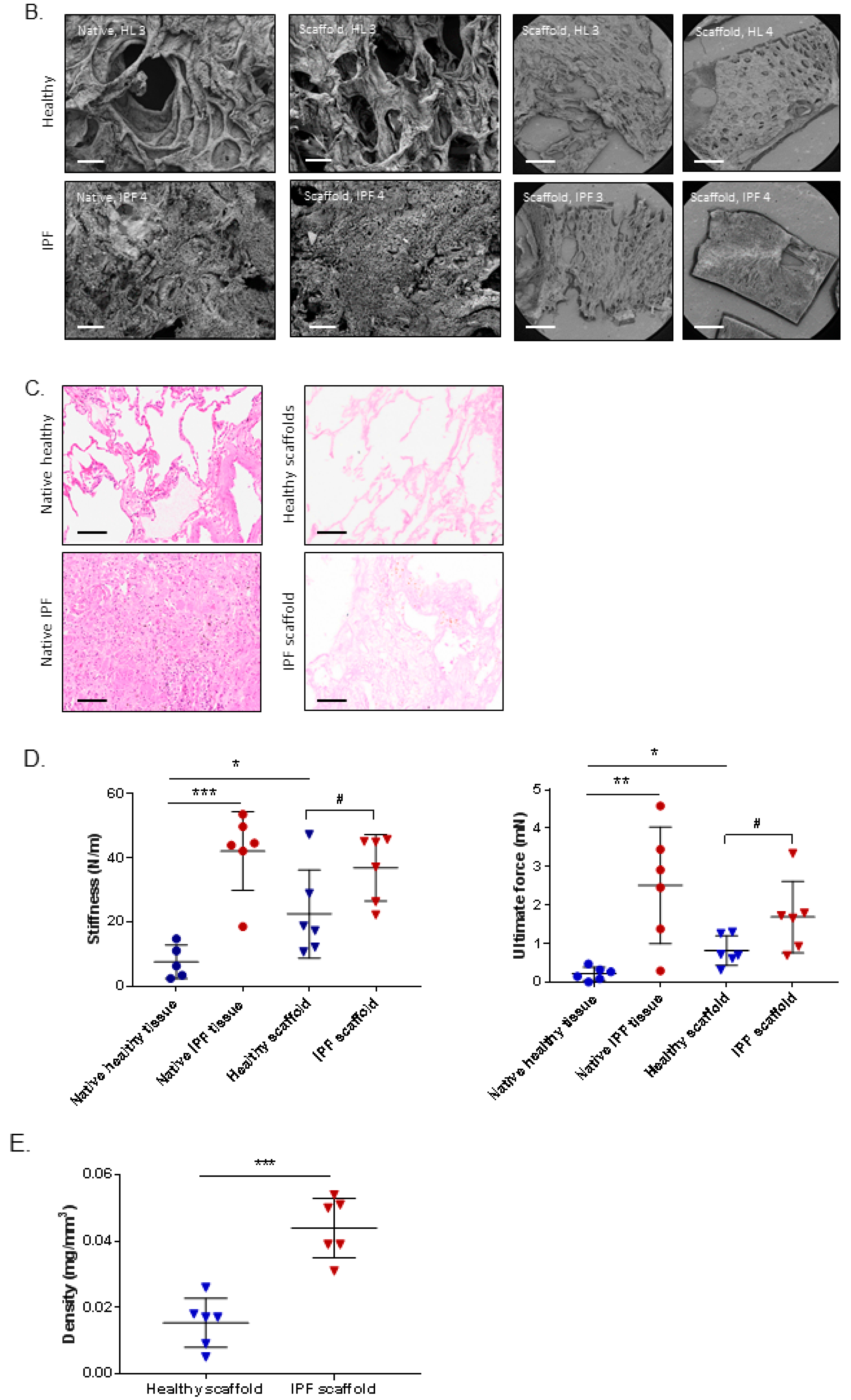
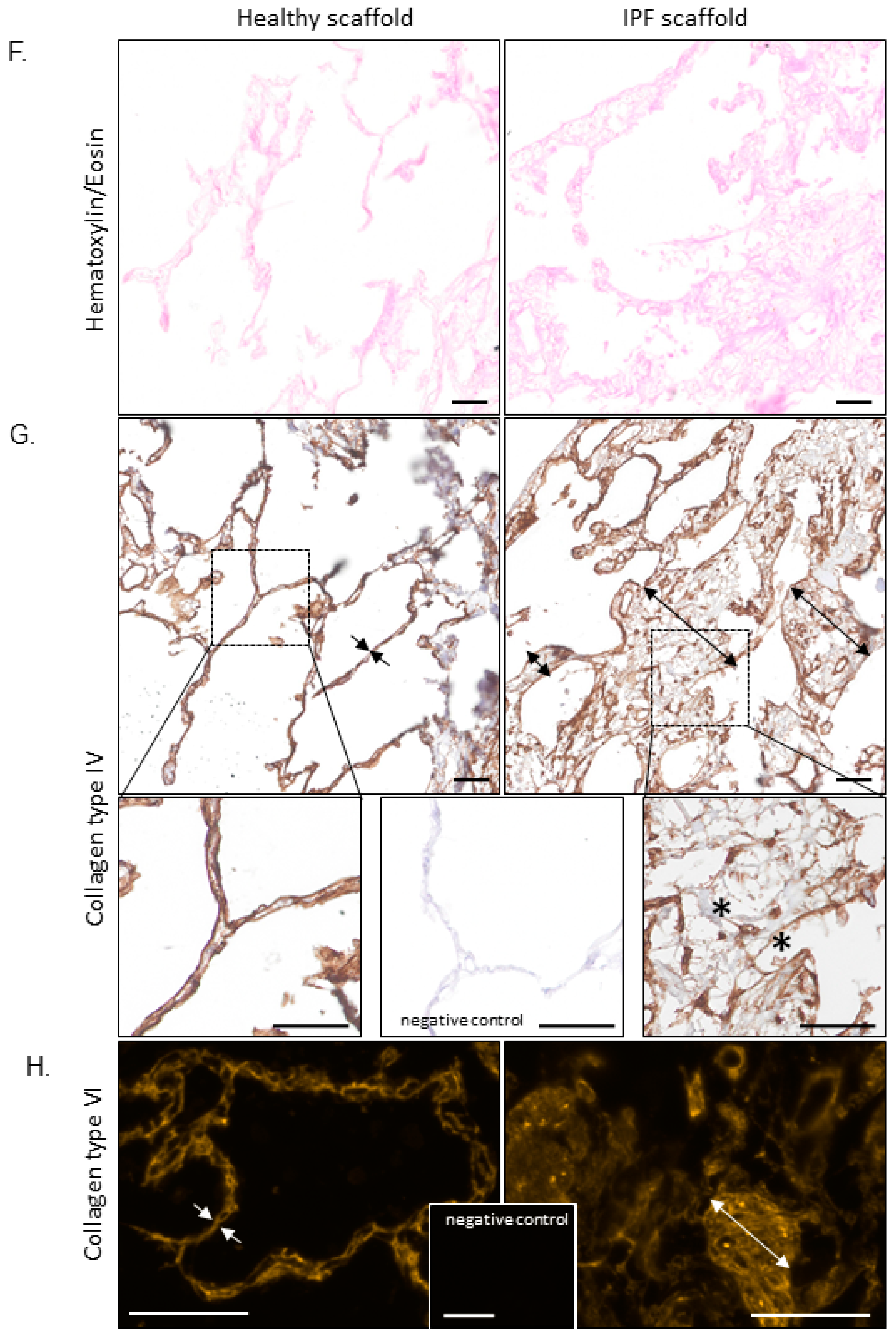
2.1. Morphology and Biomechanical Properties of Native Tissue and Scaffolds Derived from Healthy and IPF Lung Tissue
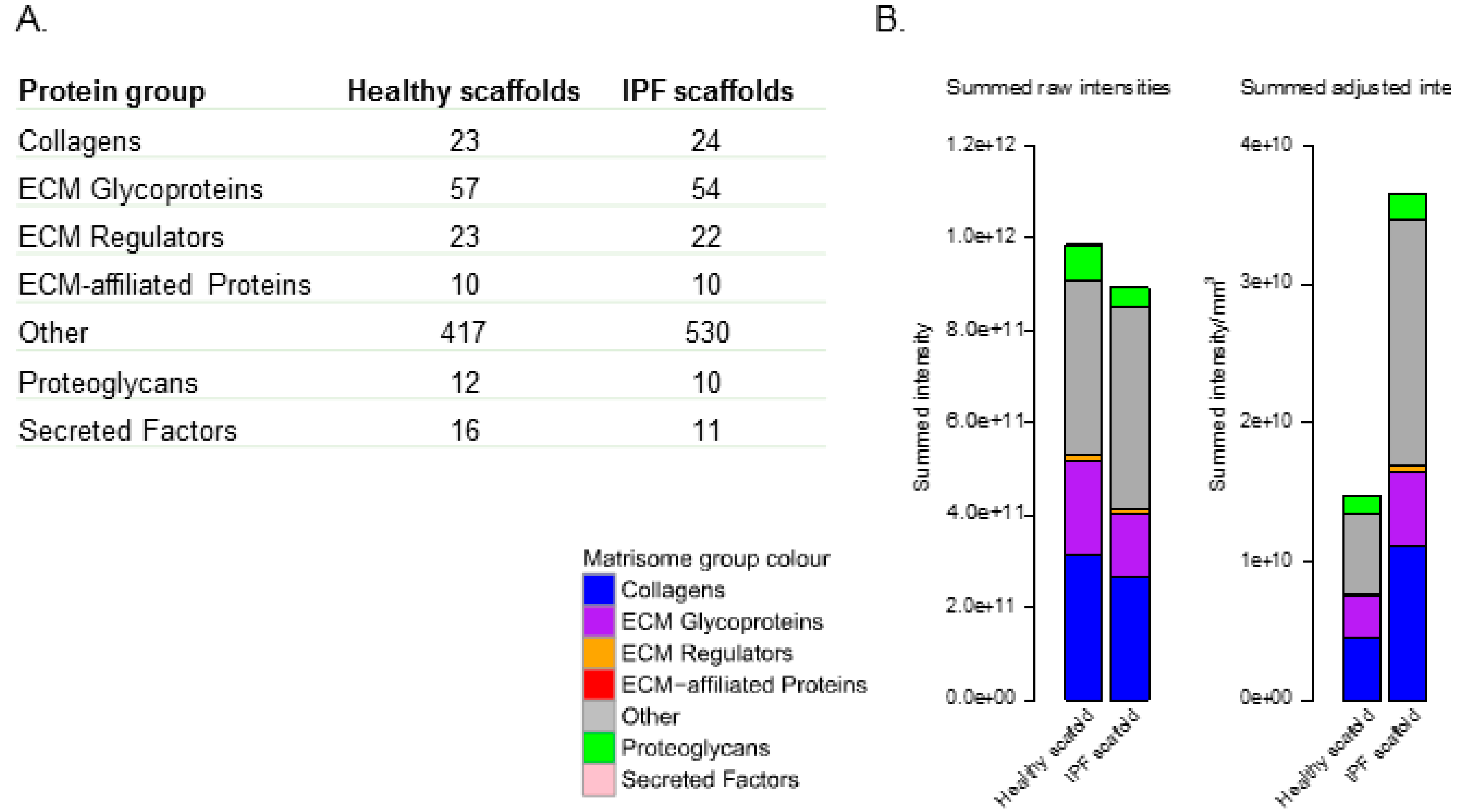
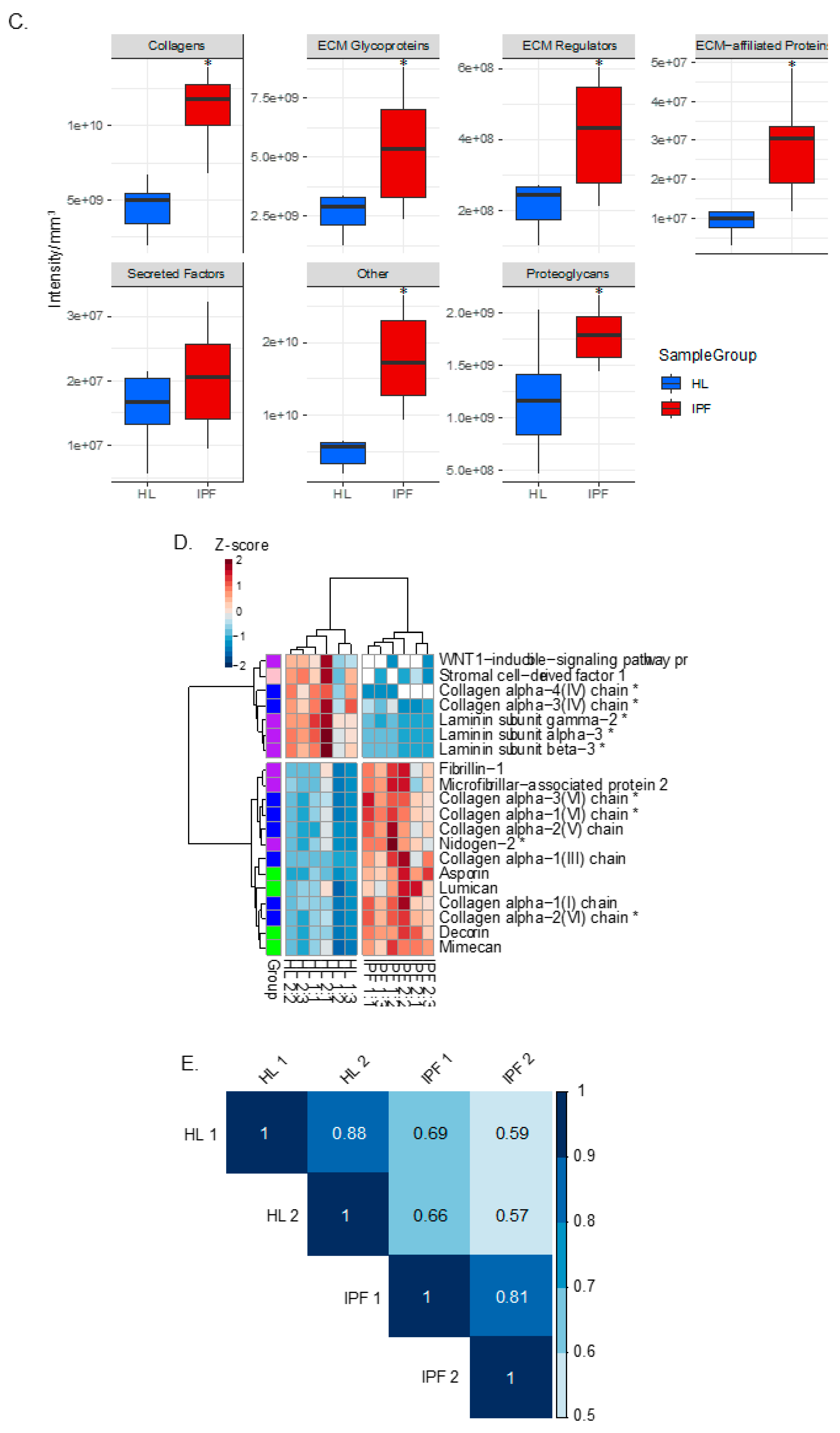
2.2. Proteomic Profiling of Lung Scaffolds
2.3. Repopulated Scaffolds
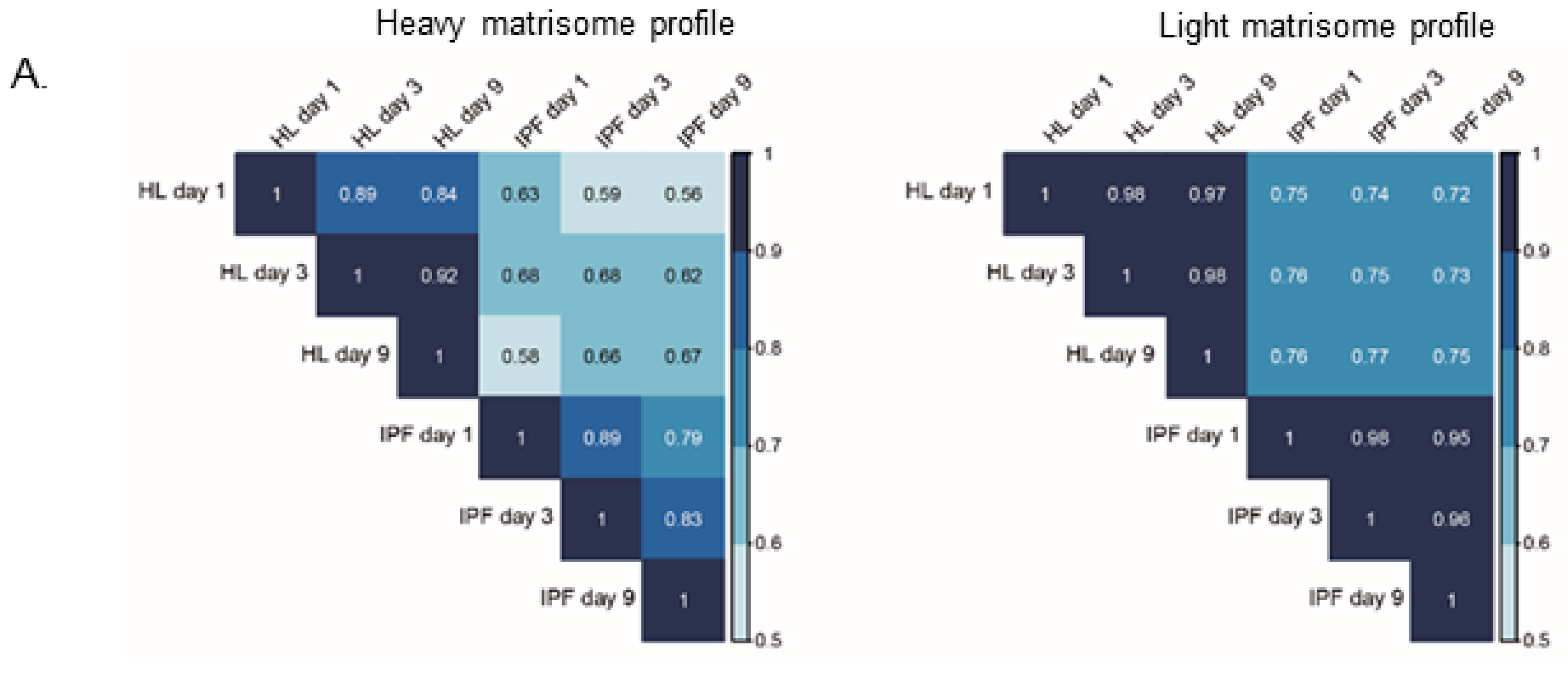
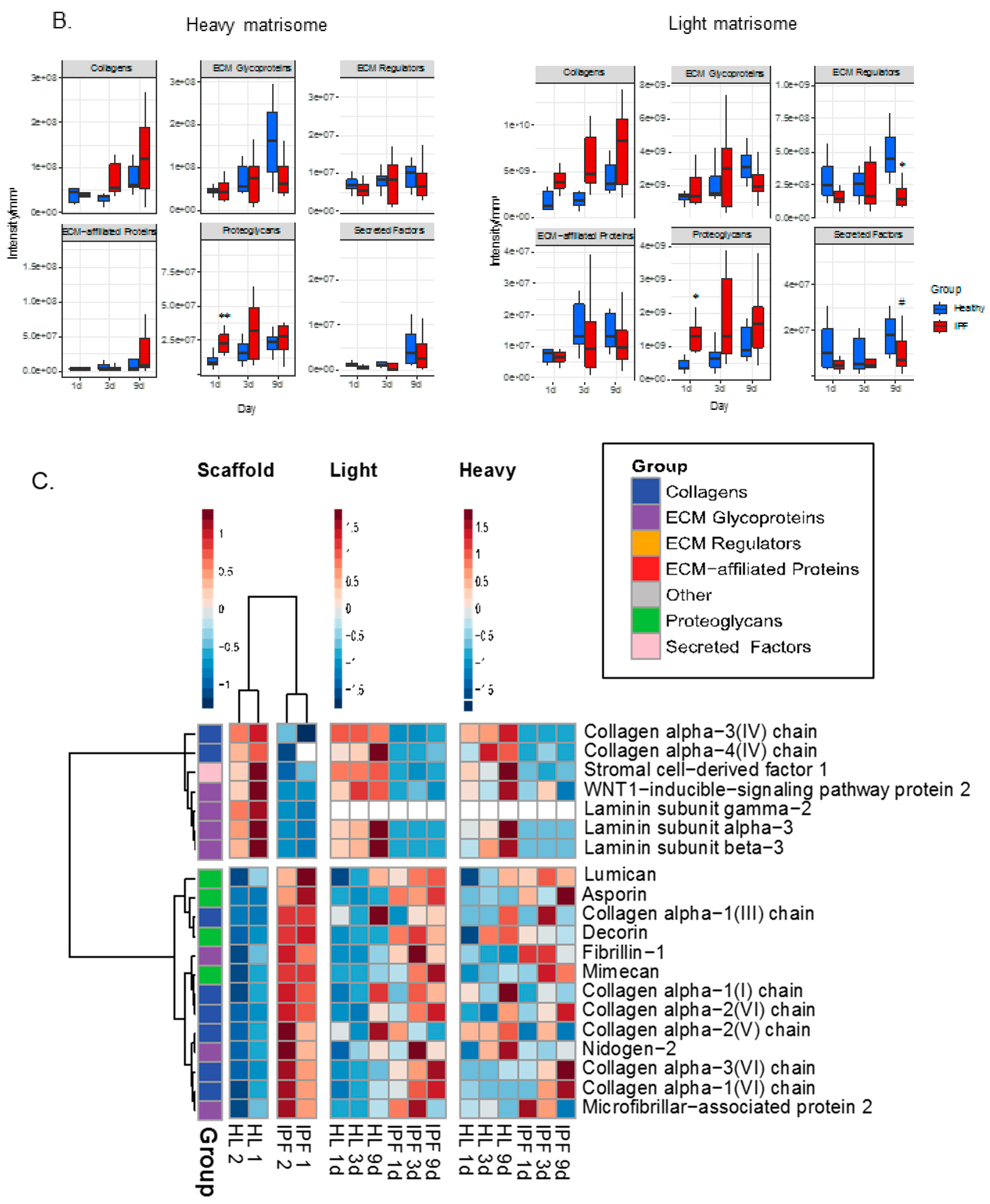
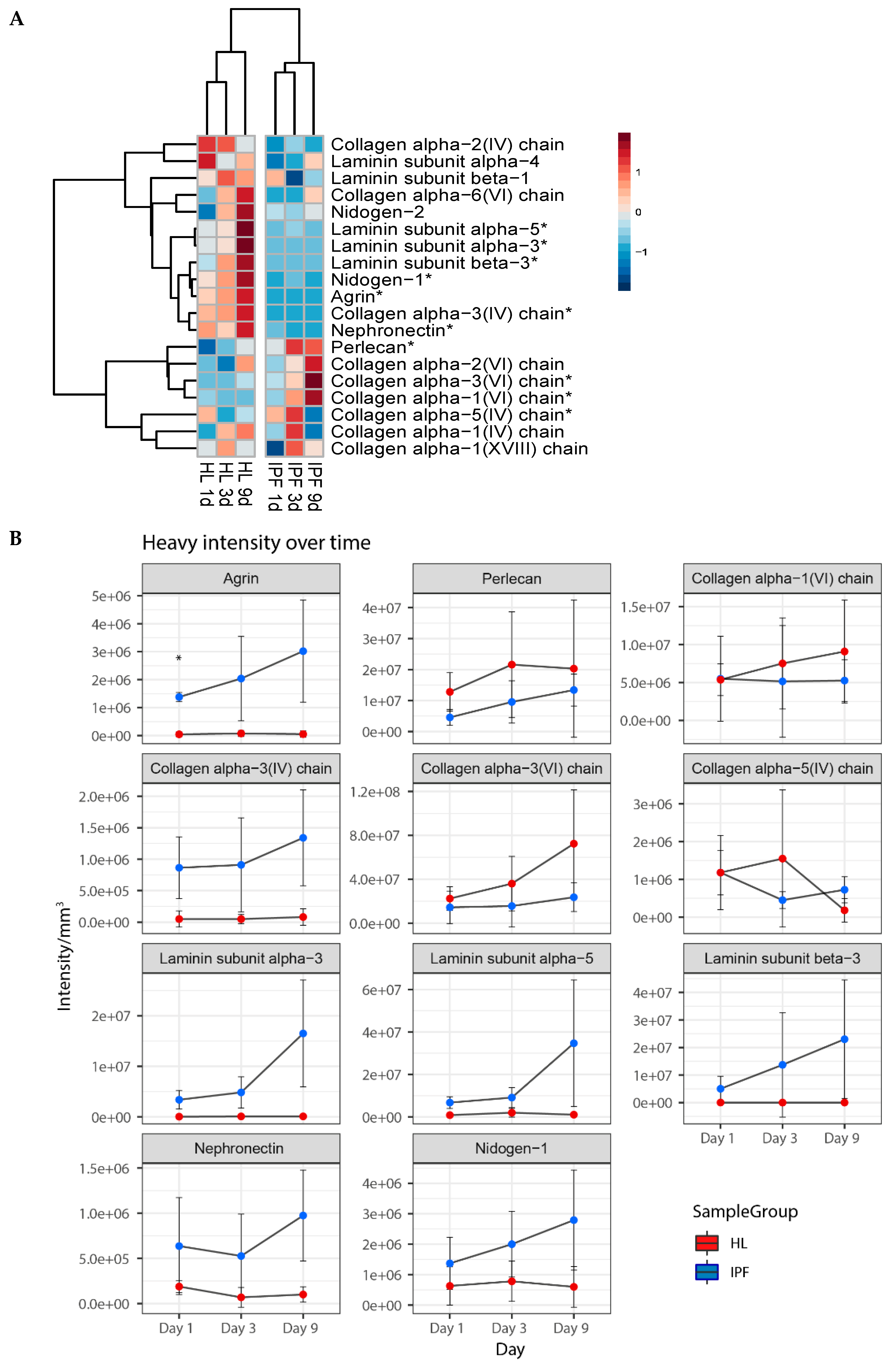
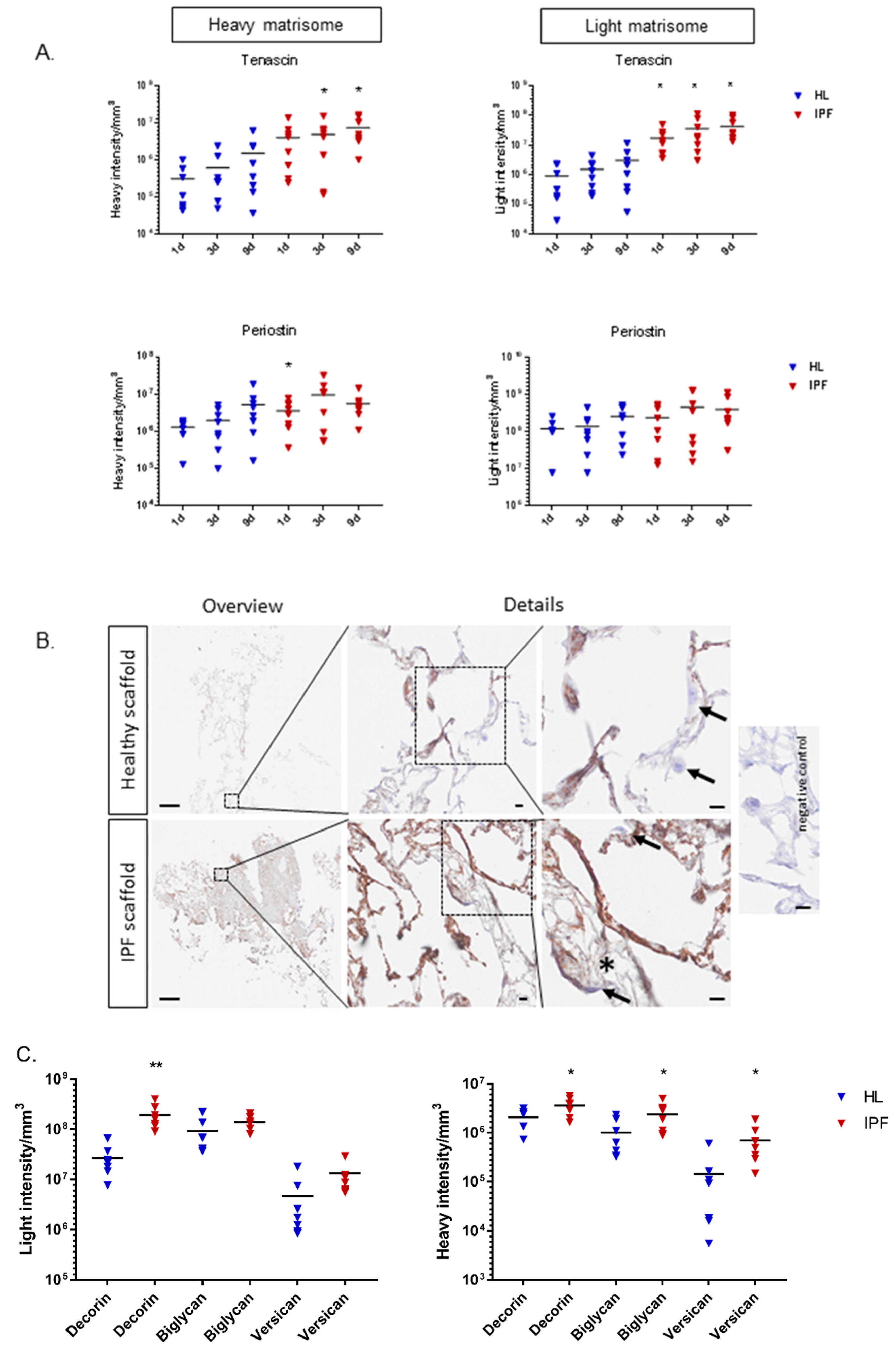
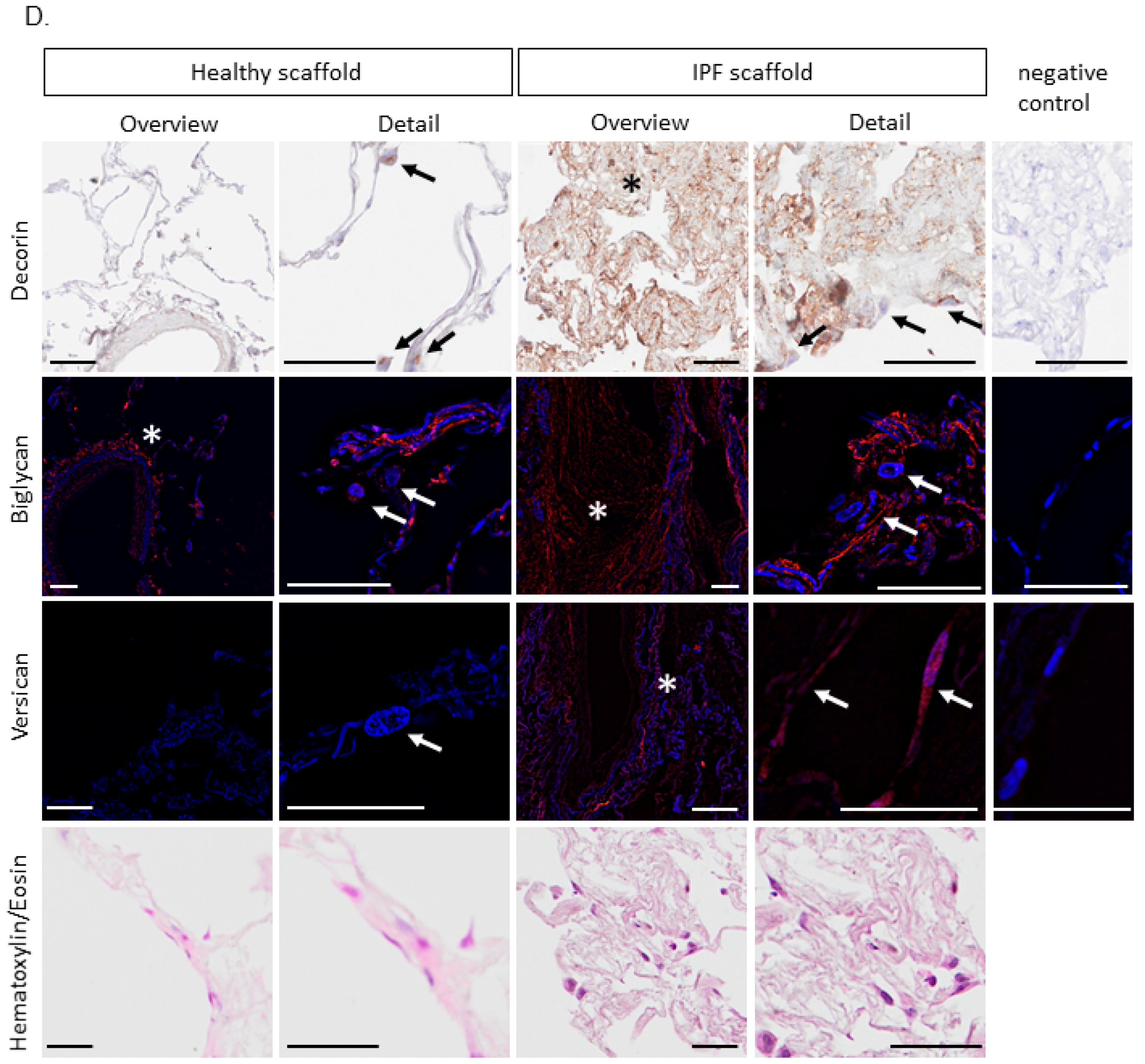
2.4. Proteomic Profiling of Matrisome Proteins in Repopulated Healthy and IPF Lung Scaffolds
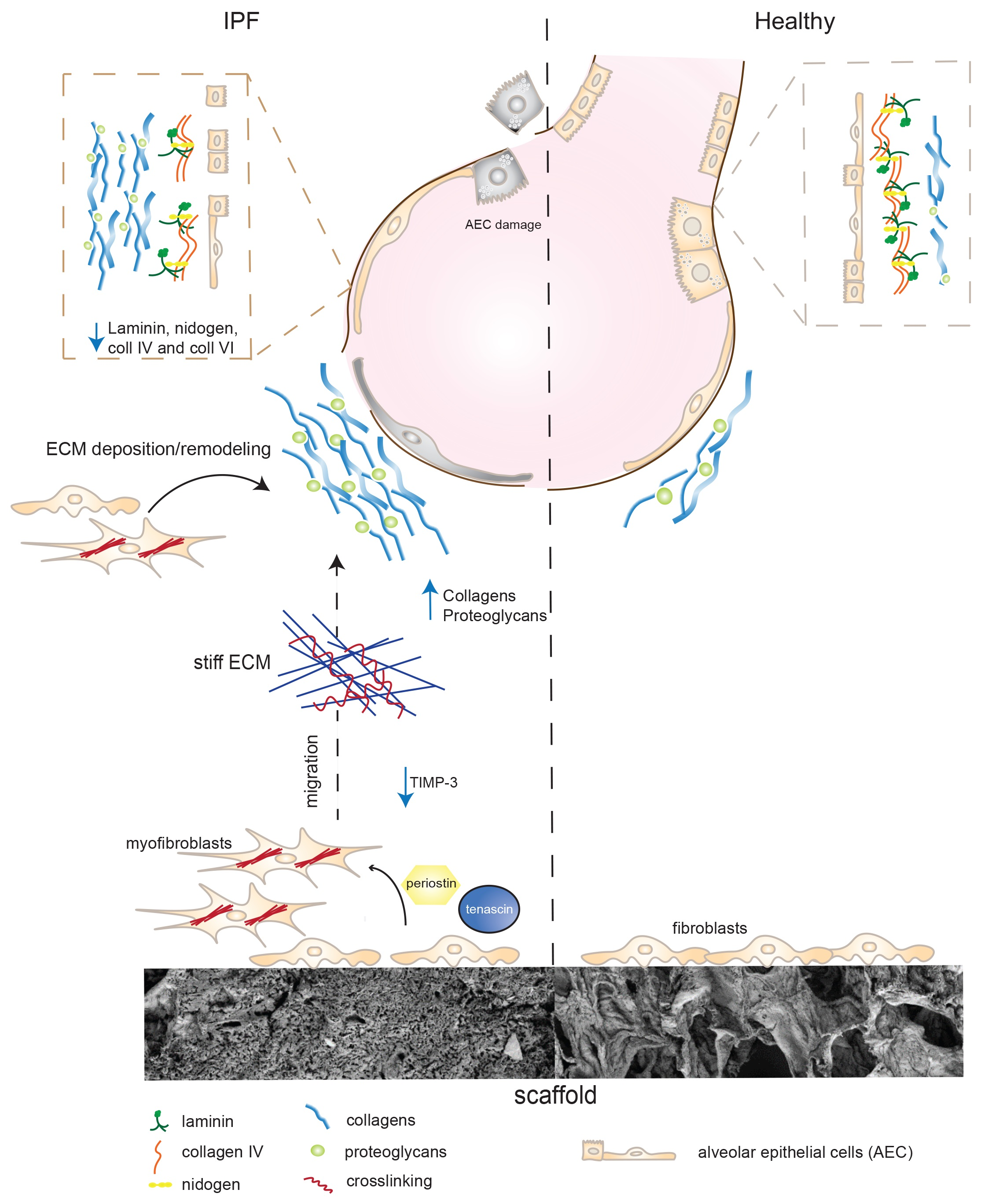
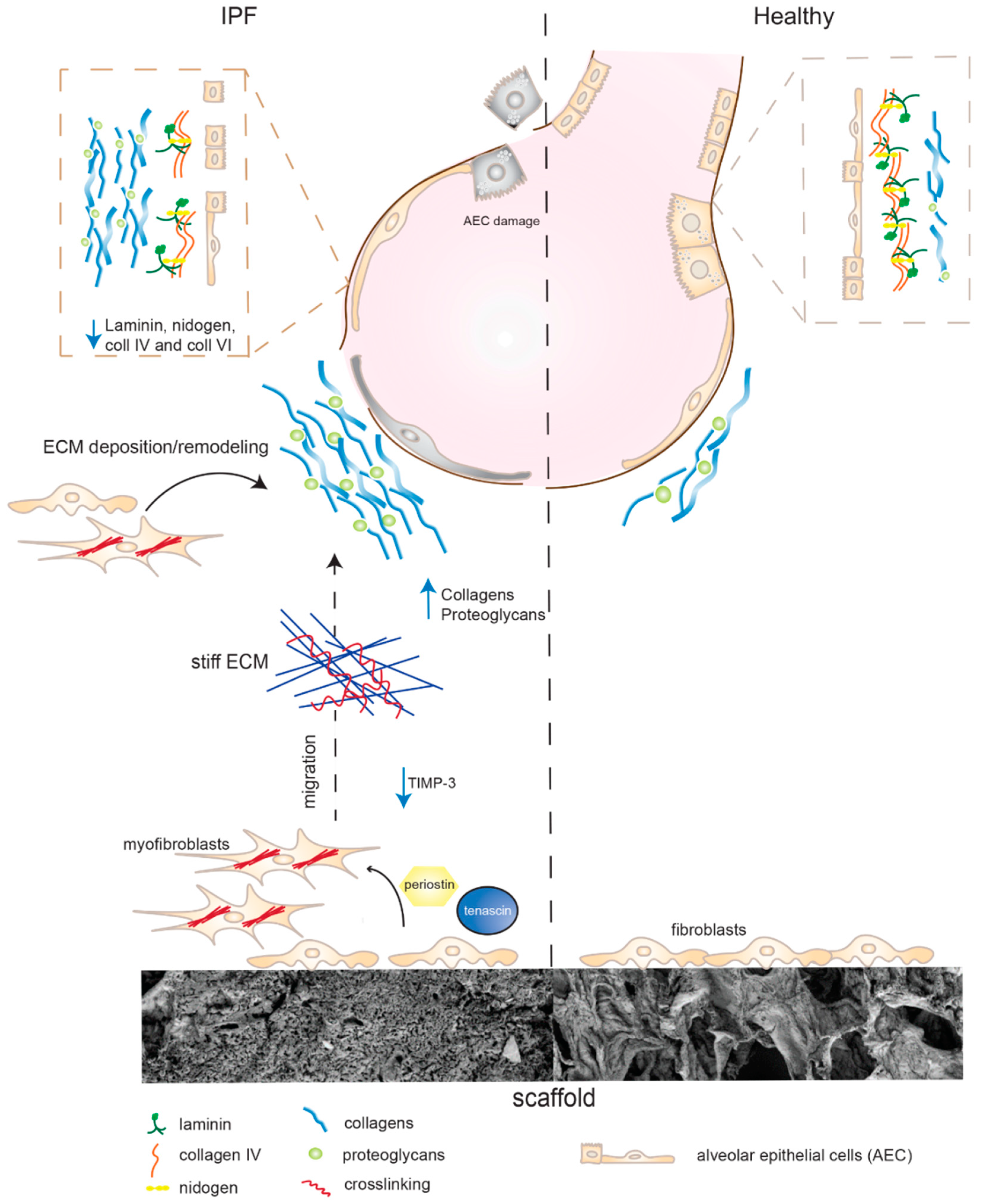
3. Discussion
4. Materials and Methods
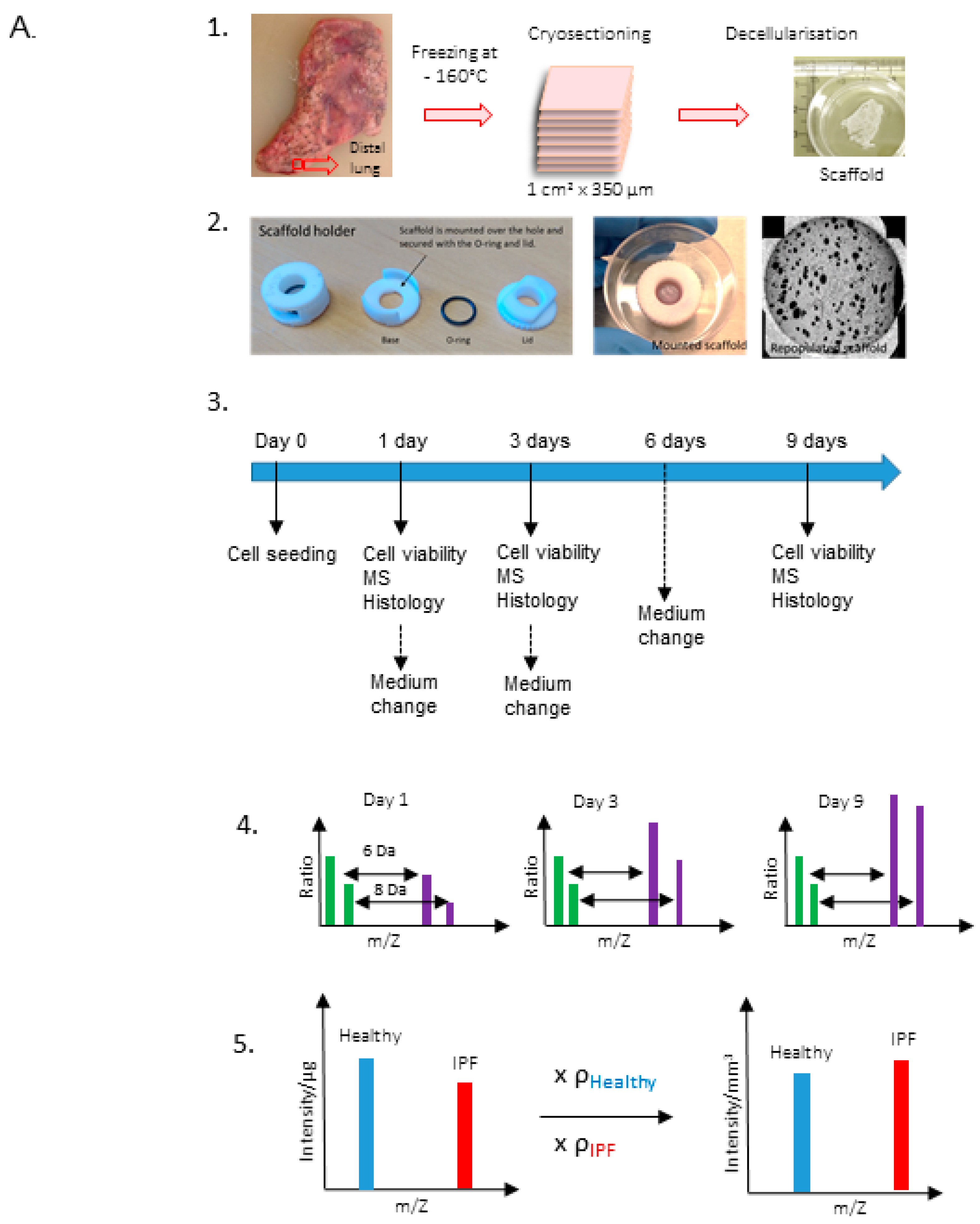
4.1. Decellularization of Lung Tissue Samples
4.2. Study Approval
4.3. DNA Measurements
4.4. Tissue Density Measurements
4.5. Mechanical Testing
4.6. Repopulation of Scaffolds with Primary Lung Fibroblasts Labeled with Heavy Arginine and Lysine
4.7. LC-MS/MS Analysis
Data Analysis
4.8. Imaging
4.8.1. Scanning Electron Microscopy (SEM)
4.8.2. Confocal Imaging
4.8.3. Fixation, Paraffin Embedding, and Sectioning
4.8.4. Hematoxylin/Eosin Staining
4.8.5. Immunohistochemistry (IHC), Immunofluorescence (IF)
4.8.6. Image Acquisition
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Darby, I.A.; Laverdet, B.; Bonte, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [PubMed] [Green Version]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [PubMed]
- Andersson-Sjoland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramirez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, S.; Ito, S.; Takahashi, K.; Furuya, K.; Kondo, M.; Sokabe, M.; Hasegawa, Y. Matrix stiffness regulates migration of human lung fibroblasts. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, J.L.; Niklason, L.E. Extracellular matrix as a driver for lung regeneration. Ann. Biomed. Eng. 2015, 43, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Simpson, J.K.; Saini, G.; Bentley, J.H.; Russell, A.M.; Braybrooke, R.; Molyneaux, P.L.; McKeever, T.M.; Wells, A.U.; Flynn, A.; et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: An analysis from the prospective, multicentre PROFILE study. Lancet Respir. Med. 2015, 3, 462–472. [Google Scholar] [CrossRef]
- Tian, Y.; Li, H.; Gao, Y.; Liu, C.; Qiu, T.; Wu, H.; Cao, M.; Zhang, Y.; Ding, H.; Chen, J.; et al. Quantitative proteomic characterization of lung tissue in idiopathic pulmonary fibrosis. Clin. Proteom. 2019, 16, 6. [Google Scholar] [CrossRef]
- Ahrman, E.; Hallgren, O.; Malmstrom, L.; Hedstrom, U.; Malmstrom, A.; Bjermer, L.; Zhou, X.H.; Westergren-Thorsson, G.; Malmstrom, J. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J. Proteomics 2018. [Google Scholar]
- Spagnolo, P.; Tzouvelekis, A.; Bonella, F. The Management of Patients With Idiopathic Pulmonary Fibrosis. Front. Med. (Lausanne) 2018, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Rosmark, O.; Ahrman, E.; Muller, C.; Elowsson Rendin, L.; Eriksson, L.; Malmstrom, A.; Hallgren, O.; Larsson-Callerfelt, A.K.; Westergren-Thorsson, G.; Malmstrom, J. Quantifying extracellular matrix turnover in human lung scaffold cultures. Sci. Rep. 2018, 8, 5409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Macdonald, B.; Kalluri, R. Structure and function of basement membranes. Exp. Biol. Med. (Maywood) 2007, 232, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Lagares, D. Matrix Stiffness: The Conductor of Organ Fibrosis. Curr. Rheumatol. Rep. 2018, 20, 2. [Google Scholar] [CrossRef]
- Smithmyer, M.E.; Sawicki, L.A.; Kloxin, A.M. Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease. Biomater. Sci. 2014, 2, 634–650. [Google Scholar] [CrossRef]
- Jaffar, J.; Yang, S.H.; Kim, S.Y.; Kim, H.W.; Faiz, A.; Chrzanowski, W.; Burgess, J.K. Greater cellular stiffness in fibroblasts from patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L59–L65. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Calle, E.A.; Hill, R.C.; Leiby, K.L.; Le, A.V.; Gard, A.L.; Madri, J.A.; Hansen, K.C.; Niklason, L.E. Targeted proteomics effectively quantifies differences between native lung and detergent-decellularized lung extracellular matrices. Acta Biomater. 2016, 46, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenstein, A.L.; Mukhopadhyay, S.; Myers, J.L. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum. Pathol. 2008, 39, 1275–1294. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. Faseb. J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Nicosia, J.; Larouche, J.; Zhang, Y.; Bachman, H.; Brown, A.C.; Holmgren, L.; Barker, T.H. Detection of an Integrin-Binding Mechanoswitch within Fibronectin during Tissue Formation and Fibrosis. ACS Nano 2017, 11, 7110–7117. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [PubMed] [Green Version]
- Estany, S.; Vicens-Zygmunt, V.; Llatjo, R.; Montes, A.; Penin, R.; Escobar, I.; Xaubet, A.; Santos, S.; Manresa, F.; Dorca, J.; et al. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFbeta1. BMC Pulm. Med. 2014, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell Mol. Life. Sci. 2017, 74, 4305–4314. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikstrom, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Strieter, R.M. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc. Am. Thorac. Soc. 2008, 5, 305–310. [Google Scholar] [CrossRef]
- Petersen, T.H.; Calle, E.A.; Colehour, M.B.; Niklason, L.E. Matrix composition and mechanics of decellularized lung scaffolds. Cells Tissues Organs 2012, 195, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Nischt, R.; Schmidt, C.; Mirancea, N.; Baranowsky, A.; Mokkapati, S.; Smyth, N.; Woenne, E.C.; Stark, H.J.; Boukamp, P.; Breitkreutz, D. Lack of nidogen-1 and -2 prevents basement membrane assembly in skin-organotypic coculture. J. Investig. Derm. 2007, 127, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Morales-Nebreda, L.I.; Rogel, M.R.; Eisenberg, J.L.; Hamill, K.J.; Soberanes, S.; Nigdelioglu, R.; Chi, M.; Cho, T.; Radigan, K.A.; Ridge, K.M.; et al. Lung-specific loss of alpha3 laminin worsens bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Carrino, D.A.; Lauer, M.E.; Vasanji, A.; Wylie, J.D.; Nelson, C.M.; Apte, S.S. Pericellular versican regulates the fibroblast-myofibroblast transition: A role for ADAMTS5 protease-mediated proteolysis. J. Biol. Chem. 2011, 286, 34298–34310. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [CrossRef]
- Bensadoun, E.S.; Burke, A.K.; Hogg, J.C.; Roberts, C.R. Proteoglycan deposition in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 1819–1828. [Google Scholar] [CrossRef]
- Westergren-Thorsson, G.; Hedstrom, U.; Nybom, A.; Tykesson, E.; Ahrman, E.; Hornfelt, M.; Maccarana, M.; van Kuppevelt, T.H.; Dellgren, G.; Wildt, M.; et al. Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Intj. Biochem. Cell Biol. 2017, 83, 27–38. [Google Scholar] [CrossRef]
- Westergren-Thorsson, G.; Sime, P.; Jordana, M.; Gauldie, J.; Sarnstrand, B.; Malmstrom, A. Lung fibroblast clones from normal and fibrotic subjects differ in hyaluronan and decorin production and rate of proliferation. Intj. Biochem. Cell Biol. 2004, 36, 1573–1584. [Google Scholar] [CrossRef]
- Vogel, V.; Sheetz, M. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef]
- Murray, M.E.; Mendez, M.G.; Janmey, P.A. Substrate stiffness regulates solubility of cellular vimentin. Mol. Biol. Cell 2014, 25, 87–94. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Haak, A.J.; Tan, Q.; Tschumperlin, D.J. Matrix biomechanics and dynamics in pulmonary fibrosis. Matrix Biol. 2018, 73, 64–76. [Google Scholar] [CrossRef]
- Melo, E.; Cardenes, N.; Garreta, E.; Luque, T.; Rojas, M.; Navajas, D.; Farre, R. Inhomogeneity of local stiffness in the extracellular matrix scaffold of fibrotic mouse lungs. J. Mech. Behav. Biomed. Mater. 2014, 37, 186–195. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.; Richter, A.G.; O’Kane, C.; McAuley, D.F.; Thickett, D.R. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J. 2009, 33, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef]
- Trebaul, A.; Chan, E.K.; Midwood, K.S. Regulation of fibroblast migration by tenascin-C. Biochem. Soc. Trans. 2007, 35, 695–697. [Google Scholar] [CrossRef]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef]
- Hallgren, O.; Nihlberg, K.; Dahlback, M.; Bjermer, L.; Eriksson, L.T.; Erjefalt, J.S.; Lofdahl, C.G.; Westergren-Thorsson, G. Altered fibroblast proteoglycan production in COPD. Respir. Res. 2010, 11, 55. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | HL 1 | HL 2 | HL 3 | HL 4 | IPF 1 | IPF 2 | IPF 3 | IPF 4 |
|---|---|---|---|---|---|---|---|---|
| Group | Healthy | Healthy | Healthy | Healthy | IPF | IPF | IPF | IPF |
| Age | 55 | 41 | 62 | 86 | 61 | 57 | 62 | 68 |
| Gender | Female | Female | Male | Male | Female | Female | Female | Male |
| Smoking history | Yes | No | No | No | Yes | Yes | Yes | Yes |
| Non-lung disease | Alpha 1 anti-trypsin deficiency | |||||||
| Lung disease | No | No | No | Squamous cell carcinoma * | IPF | IPF | IPF | IPF |
| Antibody | Protein Group | Catalogue Number | Dilution | HIER | Method | Secondary Antibody |
|---|---|---|---|---|---|---|
| Collagen type IV (α1/α2) | Collagens | Abcam, ab6586 | 1:4000 | low pH | IHC | HRP-coupled |
| Collagen type VI (α1) | Abcam, ab6588 | 1:1000 | low pH | IF | A-21246 | |
| Decorin | Proteoglycans | Atlas Antibodies, HPA003315 | 1:1000 | high pH | IHC | HRP-coupled |
| Biglycan | Atlas Antibodies, HPA003157 | 1:500 | high pH | IF | A-21246 | |
| Versican | Atlas Antibodies, HPA004726 | 1:500 | high pH | IF | A-21246 | |
| Periostin | Glycoprotein | Abcam, ab79946 | 1:1000 | low pH | IHC | HRP-coupled |
| Vimentin | Cytoskeleton | R&D, AF2105 | 1:200 | low pH | IF | A-21432 |
| Vinculin | Focal adhesions | Sigma-Aldrich, HPA063777 | 1:100 | low pH | IF | A-21246 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elowsson Rendin, L.; Löfdahl, A.; Åhrman, E.; Müller, C.; Notermans, T.; Michaliková, B.; Rosmark, O.; Zhou, X.-H.; Dellgren, G.; Silverborn, M.; et al. Matrisome Properties of Scaffolds Direct Fibroblasts in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2019, 20, 4013. https://doi.org/10.3390/ijms20164013
Elowsson Rendin L, Löfdahl A, Åhrman E, Müller C, Notermans T, Michaliková B, Rosmark O, Zhou X-H, Dellgren G, Silverborn M, et al. Matrisome Properties of Scaffolds Direct Fibroblasts in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2019; 20(16):4013. https://doi.org/10.3390/ijms20164013
Chicago/Turabian StyleElowsson Rendin, Linda, Anna Löfdahl, Emma Åhrman, Catharina Müller, Thomas Notermans, Barbora Michaliková, Oskar Rosmark, Xiao-Hong Zhou, Göran Dellgren, Martin Silverborn, and et al. 2019. "Matrisome Properties of Scaffolds Direct Fibroblasts in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 20, no. 16: 4013. https://doi.org/10.3390/ijms20164013
APA StyleElowsson Rendin, L., Löfdahl, A., Åhrman, E., Müller, C., Notermans, T., Michaliková, B., Rosmark, O., Zhou, X. -H., Dellgren, G., Silverborn, M., Bjermer, L., Malmström, A., Larsson-Callerfelt, A. -K., Isaksson, H., Malmström, J., & Westergren-Thorsson, G. (2019). Matrisome Properties of Scaffolds Direct Fibroblasts in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences, 20(16), 4013. https://doi.org/10.3390/ijms20164013