Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis


Abstract
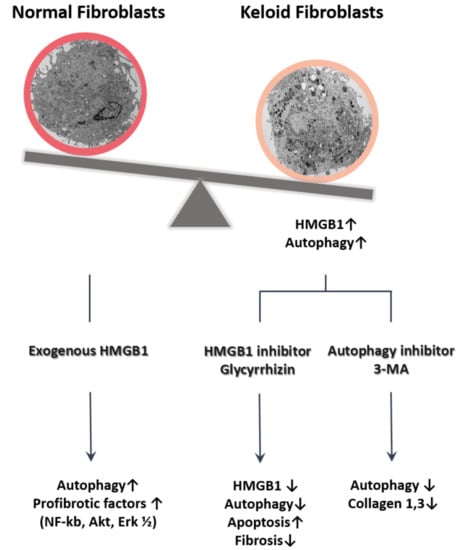
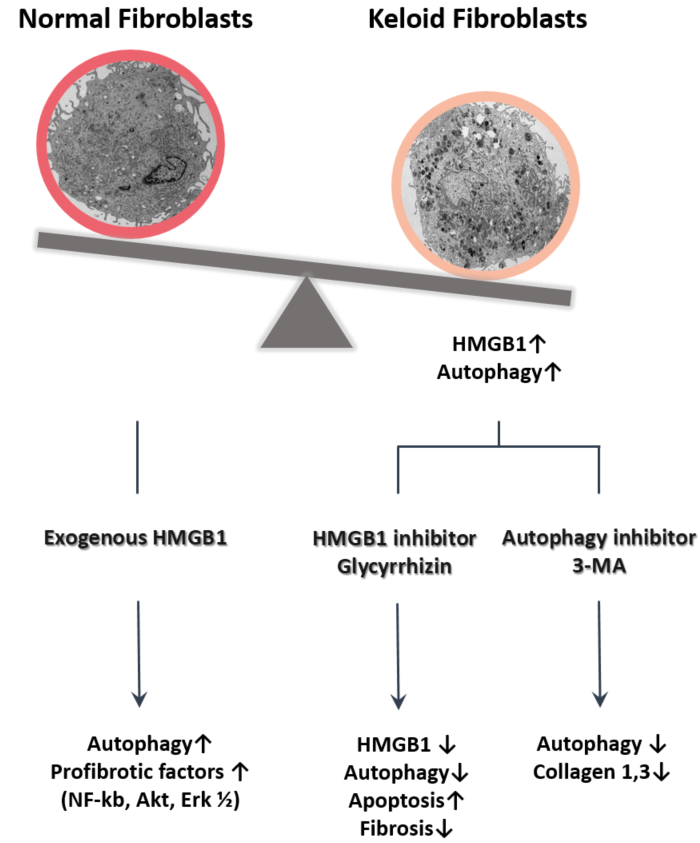
:
1. Introduction
2. Results
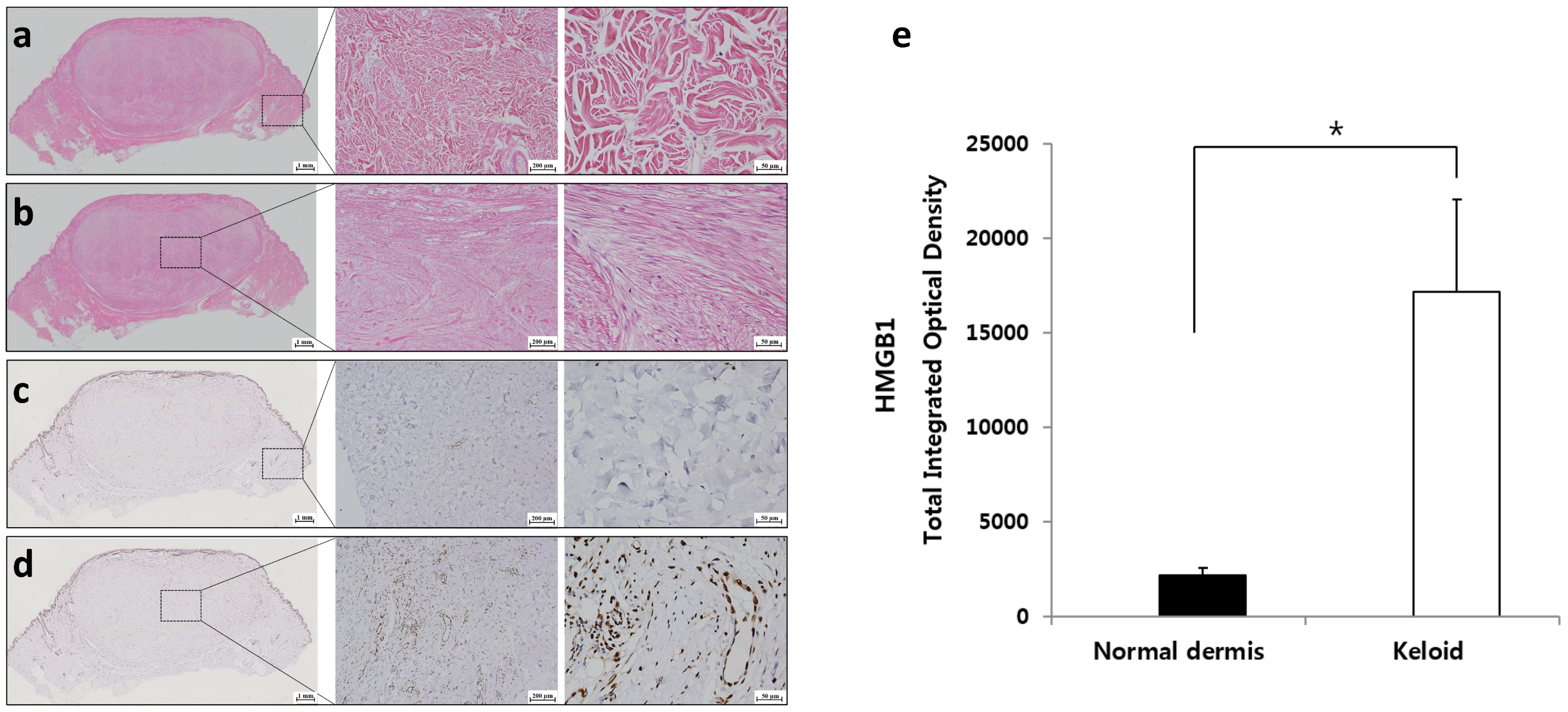
2.1. Expression of HMGB1 in Keloids
2.2. Autophagy Level in Fibrotic Condition
2.3. Effect of Glycyrrhizin on HMGB1 Expression and Cellular Viability in Keloids
2.4. Effect of Glycyrrhizin on Apoptosis and Autophagy of Keloids
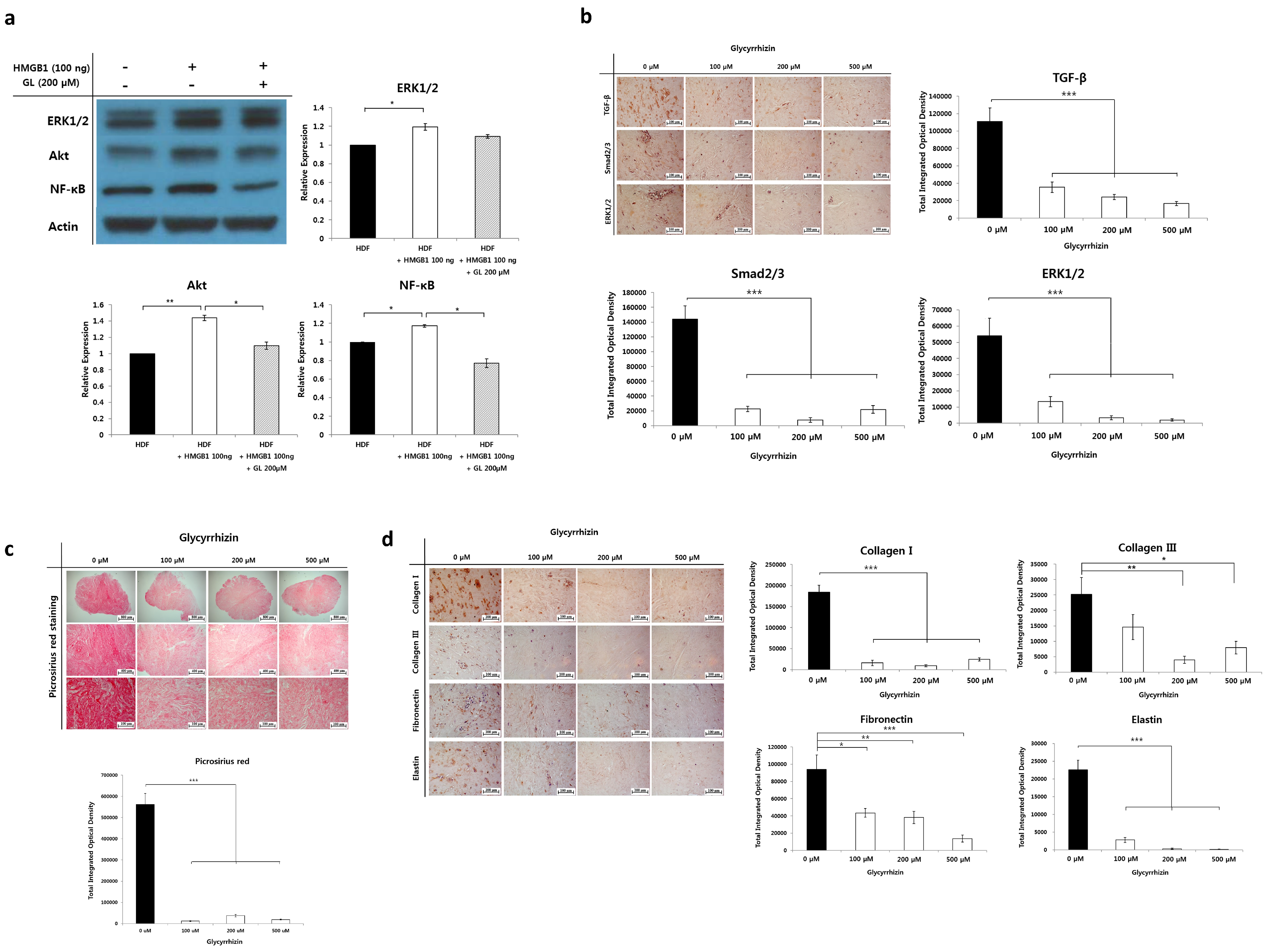
2.5. Effect of Glycyrrhizin on Profibrotic Factors, TGF-β Related Signaling Pathway, and Extracellular Matrix Components in Keloids
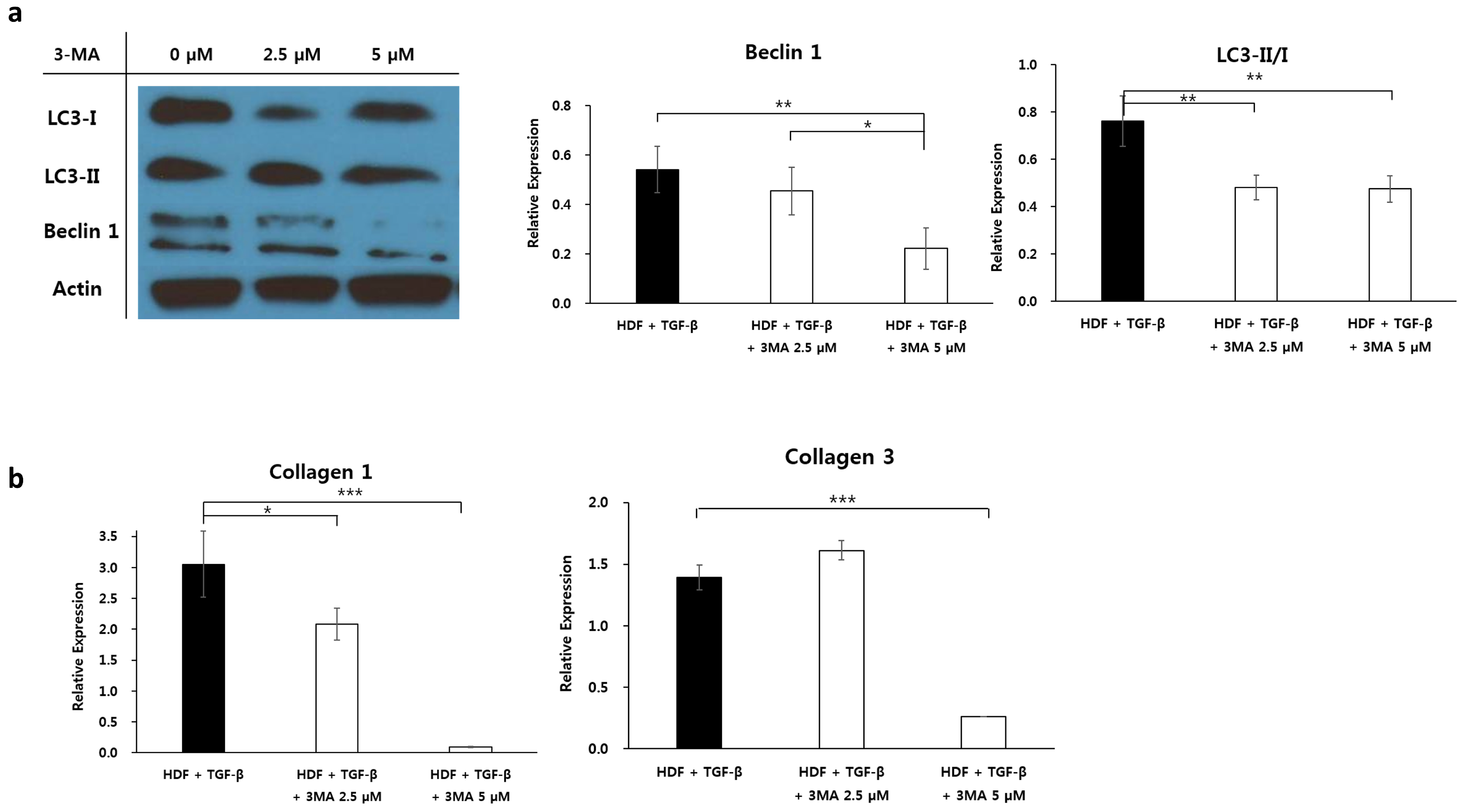
2.6. Effect of Autophagy Inhibitor on Collagen Accumulation in Fibrotic Condition
3. Discussion
4. Materials and Methods
4.1. Preparation of Cells, Tissue, and Keloid Spheroids
4.2. Histologic and Immunohistochemical Assessment
4.3. Transmission Electron Microscopy
4.4. Analysis of Cellular Viability, Autophagy, and Apoptosis
4.5. Western Blot Analysis
4.6. Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Niessen, F.B.; Spauwen, P.H.; Schalkwijk, J.; Kon, M. On the nature of hypertrophic scars and keloids: A review. Plast. Reconstr. Surg. 1999, 104, 1435–1458. [Google Scholar] [CrossRef] [PubMed]
- Yun, I.S.; Lee, M.H.; Rah, D.K.; Lew, D.H.; Park, J.C.; Lee, W.J. Heat Shock Protein 90 Inhibitor (17-AAG) Induces Apoptosis and Decreases Cell Migration/Motility of Keloid Fibroblasts. Plast. Reconstr. Surg. 2015, 136, 44e–53e. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.U.; Lee, W.J.; Tran, T.N.; Jung, I.; Lee, J.H. Hsp70 Knockdown by siRNA Decreased Collagen Production in Keloid Fibroblasts. Yonsei. Med. J. 2015, 56, 1619–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Shin, J.U.; Jung, I.; Lee, H.; Rah, D.K.; Jung, J.Y.; Lee, W.J. Proteomic profiling reveals upregulated protein expression of hsp70 in keloids. Biomed. Res. Int. 2013, 2013, 621538. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, A.; Mess, S.; Thomassen, J.M.; Kauffman, C.L.; Davison, S.P. Keloid pathogenesis and treatment. Plast. Reconstr. Surg. 2006, 117, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Bran, G.M.; Goessler, U.R.; Hormann, K.; Riedel, F.; Sadick, H. Keloids: Current concepts of pathogenesis (review). Int. J. Mol. Med. 2009, 24, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Robles, D.T.; Moore, E.; Draznin, M.; Berg, D. Keloids: Pathophysiology and management. Dermatol. Online J. 2007, 13, 9. [Google Scholar] [PubMed]
- Luo, S.; Benathan, M.; Raffoul, W.; Panizzon, R.G.; Egloff, D.V. Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plast. Reconstr. Surg. 2001, 107, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.N.; Soo, C.; Shaw, W.W.; Watson, J.; Messadi, D.; Longaker, M.T.; Zhang, X.; Ting, K. Downregulation of apoptosis-related genes in keloid tissues. J. Surg. Res. 1999, 87, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, H.; Miyauchi, S.; Miki, Y. Proliferating activity of dermal fibroblasts in keloids and hypertrophic scars. Acta Derm. -Venereol. 1995, 75, 102–104. [Google Scholar] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Et Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnanasekar, M.; Kalyanasundaram, R.; Zheng, G.; Chen, A.; Bosland, M.C.; Kajdacsy-Balla, A. HMGB1: A Promising Therapeutic Target for Prostate Cancer. Prostate Cancer 2013, 2013, 157103. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; DeMarco, R.A. Dealing with death: HMGB1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs 2003, 4, 1405–1409. [Google Scholar] [PubMed]
- Li, L.C.; Gao, J.; Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, A.; Uyanik, M.H.; Cerrah, S.; Altas, S.; Dursun, H.; Demir, M.; Uslu, H. Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol. 2010, 23, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., III; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Loze, M.T.; Zeh, H.J.; Kang, R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy 2010, 6, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Maeyama, T.; Kawaguchi, T.; Yoshimi, M.; Fukumoto, J.; Yamada, M.; Yamada, S.; Kuwano, K.; Nakanishi, Y. The role of high mobility group box1 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.T.; Yao, Q.Y.; Xu, B.L.; Wang, J.Y.; Zhou, C.H.; Zhang, S.C. Protective effects of curcumin against hepatic fibrosis induced by carbon tetrachloride: Modulation of high-mobility group box 1, Toll-like receptor 4 and 2 expression. Food Chem. Toxicol. 2012, 50, 3343–3351. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.K.; Wang, B.; Lu, Q.H.; Zhang, W.; Qin, W.D.; Liu, X.J.; Liu, X.Q.; An, F.S.; Zhang, Y.; Zhang, M.X. Inhibition of high-mobility group box 1 improves myocardial fibrosis and dysfunction in diabetic cardiomyopathy. Int. J. Cardiol. 2014, 172, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Nobili, V.; Ceccarelli, S.; Panera, N.; De Stefanis, C.; De Vito, R.; Vitali, R.; Bedogni, G.; Balsano, C.; Cucchiara, S.; et al. Plasma high mobility group box 1 protein reflects fibrosis in pediatric nonalcoholic fatty liver disease. Expert Rev. Mol. Diagn. 2014, 14, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012, 8, 275–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.H.; Li, T.; Chang, L.L.; Zhu, H.; Tong, Y.G.; Chen, X.P.; Wang, Y.T.; Lu, J.J. Glycyrrhetinic Acid triggers a protective autophagy by activation of extracellular regulated protein kinases in hepatocellular carcinoma cells. J. Agric. Food Chem. 2014, 62, 11910–11916. [Google Scholar] [CrossRef] [PubMed]
- Smolarczyk, R.; Cichon, T.; Matuszczak, S.; Mitrus, I.; Lesiak, M.; Kobusinska, M.; Kamysz, W.; Jarosz, M.; Sieron, A.; Szala, S. The role of Glycyrrhizin, an inhibitor of HMGB1 protein, in anticancer therapy. Arch. Immunol. Et Ther. Exp. 2012, 60, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.P. A direct inhibitor of HMGB1 cytokine. Chem. Biol. 2007, 14, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Katsuki, H.; Fukutomi, C.; Takahashi, M.; Motomura, M.; Fukunaga, M.; Matsuoka, Y.; Isohama, Y.; Izumi, Y.; Kume, T.; et al. HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology 2011, 61, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Ward, M.F.; Sama, A.E.; Wang, H. High mobility group box 1 protein as a potential drug target for infection- and injury-elicited inflammation. Inflamm. Allergy Drug Targets 2010, 9, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Xiang, L.; Yuan, L.; Hu, L.; Wu, W.; Cai, L.; Yin, L.; Dong, H. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS ONE 2014, 9, e89450. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Appleton, I.; Brown, N.J.; Willoughby, D.A. Apoptosis, necrosis, and proliferation: Possible implications in the etiology of keloids. Am. J. Pathol. 1996, 149, 1441–1447. [Google Scholar] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Reviews Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Hu, D.H.; Zhang, Z.F.; Bai, X.Z.; Wang, H.T.; Zhu, X.X.; Su, Y.J.; Tang, C.W. Reduced expression of microtubule-associated protein 1 light chain 3 in hypertrophic scars. Arch. Dermatol. Res. 2012, 304, 209–215. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Garbi, C.; Santoriello, M.; Santillo, A.; Wilson, R.R. Differential apoptosis markers in human keloids and hypertrophic scars fibroblasts. Mol. Cell. Biochem. 2009, 327, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kose, O.; Waseem, A. Keloids and hypertrophic scars: Are they two different sides of the same coin? Dermatol. Surg. 2008, 34, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Palumbo, R.; Bianchi, M.E. HMGB1: A signal of necrosis. Autoimmunity 2007, 40, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Jackson, P.L.; Liu, G.; Hardison, M.; Livraghi, A.; Solomon, G.M.; McQuaid, D.B.; Noerager, B.D.; Gaggar, A.; Clancy, J.P.; et al. Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am. J. Respir. Crit. Care Med. 2008, 178, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Y.; Wang, Z.; Chen, L.; Dong, X.; Nie, X. Interference with HMGB1 increases the sensitivity to chemotherapy drugs by inhibiting HMGB1-mediated cell autophagy and inducing cell apoptosis. Tumour Biol. 2015, 36, 8585–8592. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, T.T.; Zhang, P.; Xu, C.J.; Rong, Z.X.; Yan, Z.Y.; Fang, C.Y. Autophagy mediates oral submucous fibrosis. Exp. Ther. Med. 2016, 11, 1859–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmouliere, A.; Badid, C.; Bochaton-Piallat, M.L.; Gabbiani, G. Apoptosis during wound healing, fibrocontractive diseases and vascular wall injury. Int. J. Biochem. Cell Biol. 1997, 29, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Asano, Y.; Taniguchi, T.; Nakamura, K.; Saigusa, R.; Miura, S.; Toyama, T.; Takahashi, T.; Ichimura, Y.; Yoshizaki, A.; et al. Glycyrrhizin Ameliorates Fibrosis, Vasculopathy, and Inflammation in Animal Models of Systemic Sclerosis. J. Investig. Dermatol. 2017, 137, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zong, L.; Xu, M.; Dong, Y.; Lu, L. Effects of 18alpha-glycyrrhizin on TGF-beta1/Smad signaling pathway in rats with carbon tetrachloride-induced liver fibrosis. Int. J. Clin. Exp. Pathol. 2015, 8, 1292–1301. [Google Scholar] [PubMed]
- Moro, T.; Shimoyama, Y.; Kushida, M.; Hong, Y.Y.; Nakao, S.; Higashiyama, R.; Sugioka, Y.; Inoue, H.; Okazaki, I.; Inagaki, Y. Glycyrrhizin and its metabolite inhibit Smad3-mediated type I collagen gene transcription and suppress experimental murine liver fibrosis. Life Sci. 2008, 83, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, D.A.; Yager, D.R.; Diegelmann, R.F.; Cohen, I.K. The effect of TGF-beta on keloid fibroblast proliferation and collagen synthesis. Plast. Reconstr. Surg. 1996, 98, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Tsujita-Kyutoku, M.; Uehara, N.; Matsuoka, Y.; Kyutoku, S.; Ogawa, Y.; Tsubura, A. Comparison of transforming growth factor-beta/Smad signaling between normal dermal fibroblasts and fibroblasts derived from central and peripheral areas of keloid lesions. In Vivo 2005, 19, 959–963. [Google Scholar] [PubMed]
- Wang, W.; Qu, M.; Xu, L.; Wu, X.; Gao, Z.; Gu, T.; Zhang, W.; Ding, X.; Liu, W.; Chen, Y.L. Sorafenib exerts an anti-keloid activity by antagonizing TGF-beta/Smad and MAPK/ERK signaling pathways. J. Mol. Med. 2016, 94, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, Z.; Shi, Y.; Sun, Y.; Lin, Z.; Jiang, H.; Hou, T.; Wang, Q.; Yuan, X.; Zhu, X.; et al. Inhibition of Smad3 expression decreases collagen synthesis in keloid disease fibroblasts. J. Plast. Reconstr. Aesthetic Surg. 2007, 60, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Choi, I.K.; Lee, J.H.; Kim, Y.O.; Yun, C.O. A novel three-dimensional model system for keloid study: Organotypic multicellular scar model. Wound Repair Regen. 2013, 21, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, J.H.; Kim, J.; Huang, J.H.; Zhang, S.N.; Kang, Y.A.; Kim, H.; Yun, C.O. Short hairpin RNA-expressing oncolytic adenovirus-mediated inhibition of IL-8: Effects on antiangiogenesis and tumor growth inhibition. Gene Ther. 2008, 15, 635–651. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Race | Age (Years) | Origin | |
|---|---|---|---|---|
| 1 | F | Korean | 18 | Ankle |
| 2 | F | Korean | 3 | Earlobe |
| 3 | M | Korean | 4 | Neck |
| 4 | M | Korean | 31 | Neck |
| 5 | F | Korean | 11 | Knee |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, Y.R.; Roh, H.; Jung, J.H.; Ahn, H.M.; Lee, J.H.; Yun, C.-O.; Lee, W.J. Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4134. https://doi.org/10.3390/ijms20174134
Jeon YR, Roh H, Jung JH, Ahn HM, Lee JH, Yun C-O, Lee WJ. Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis. International Journal of Molecular Sciences. 2019; 20(17):4134. https://doi.org/10.3390/ijms20174134
Chicago/Turabian StyleJeon, Yeo Reum, Hyun Roh, Ji Hyuk Jung, Hyo Min Ahn, Ju Hee Lee, Chae-Ok Yun, and Won Jai Lee. 2019. "Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis" International Journal of Molecular Sciences 20, no. 17: 4134. https://doi.org/10.3390/ijms20174134
APA StyleJeon, Y. R., Roh, H., Jung, J. H., Ahn, H. M., Lee, J. H., Yun, C. -O., & Lee, W. J. (2019). Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis. International Journal of Molecular Sciences, 20(17), 4134. https://doi.org/10.3390/ijms20174134