Identification of Msp1-Induced Signaling Components in Rice Leaves by Integrated Proteomic and Phosphoproteomic Analysis



Abstract
:1. Introduction
2. Results
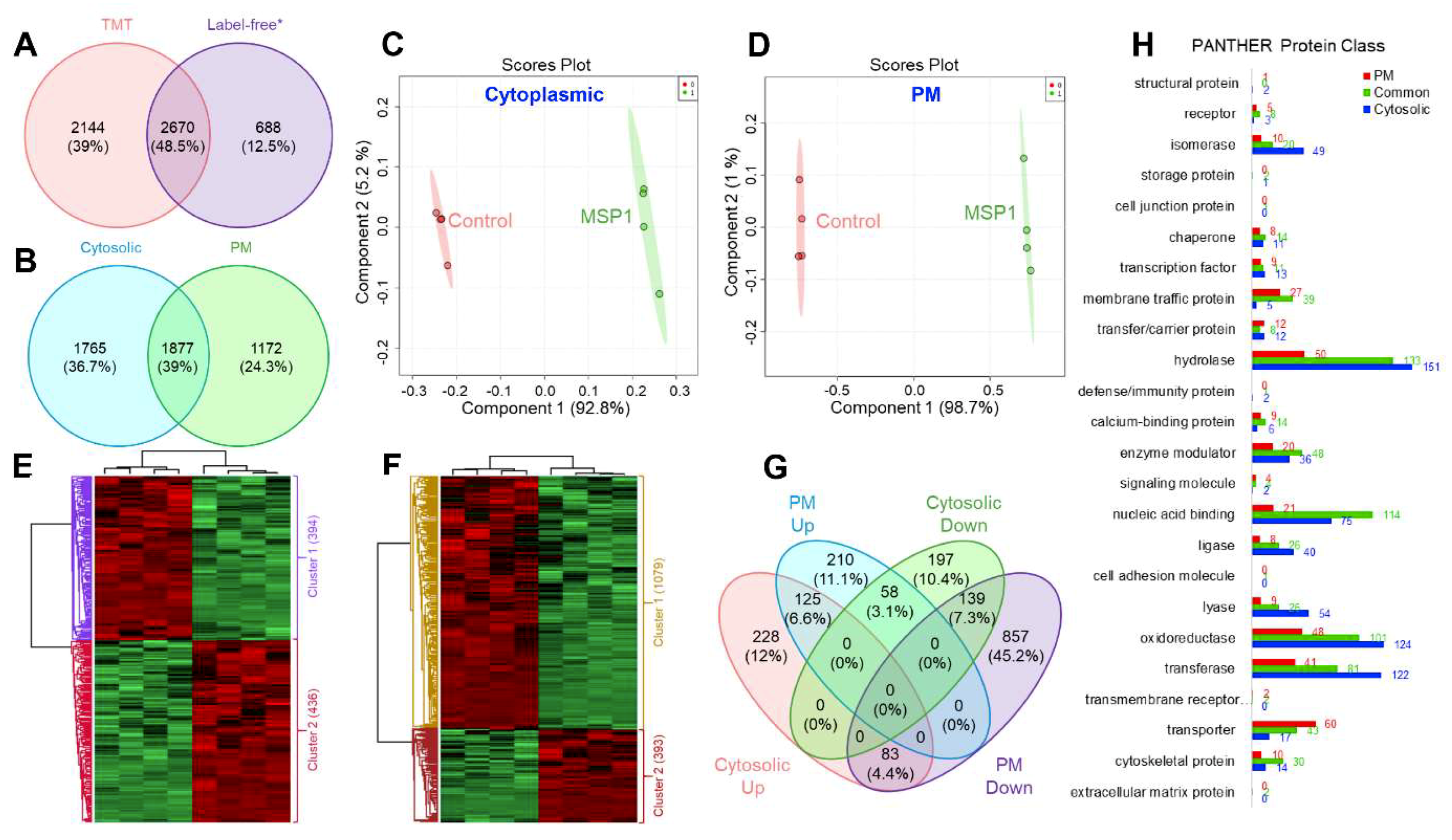
2.1. Quantitative Proteome Analysis
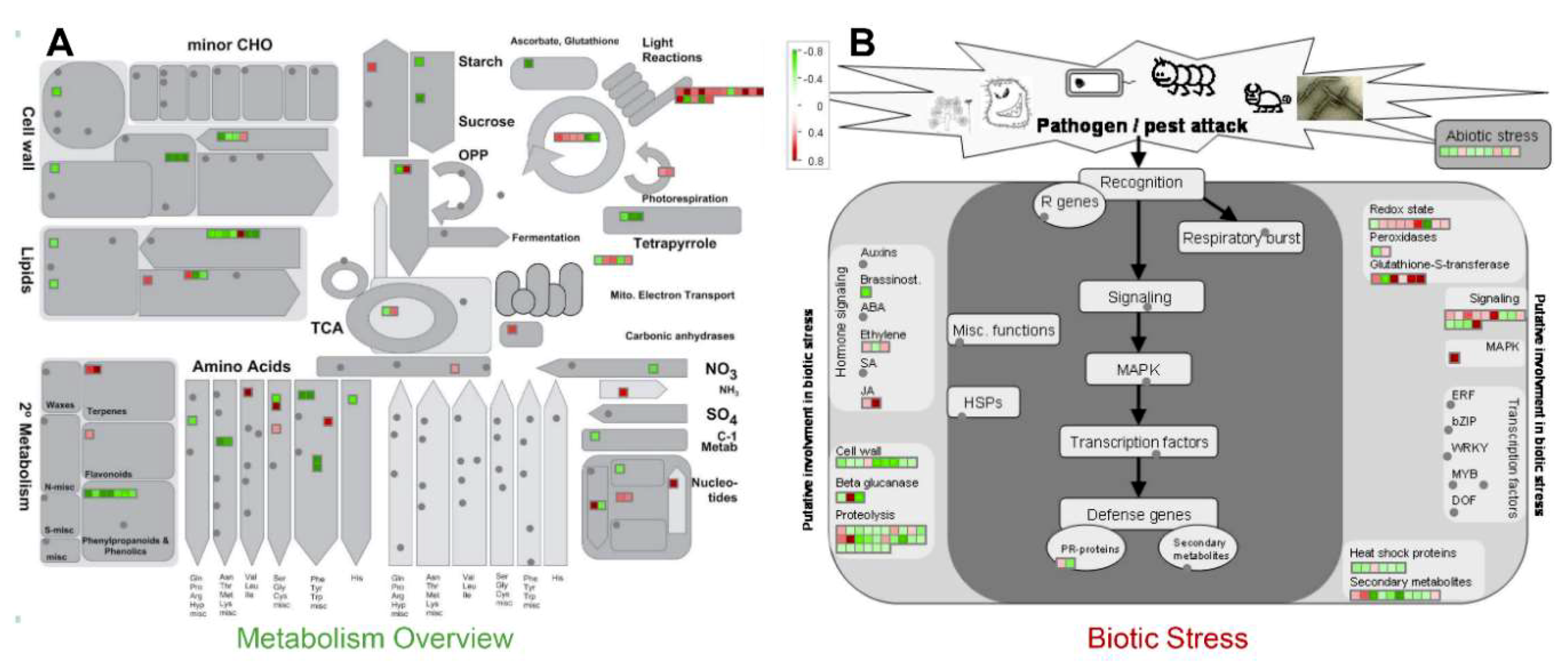
2.2. Functional Annotation of the Identified Proteins
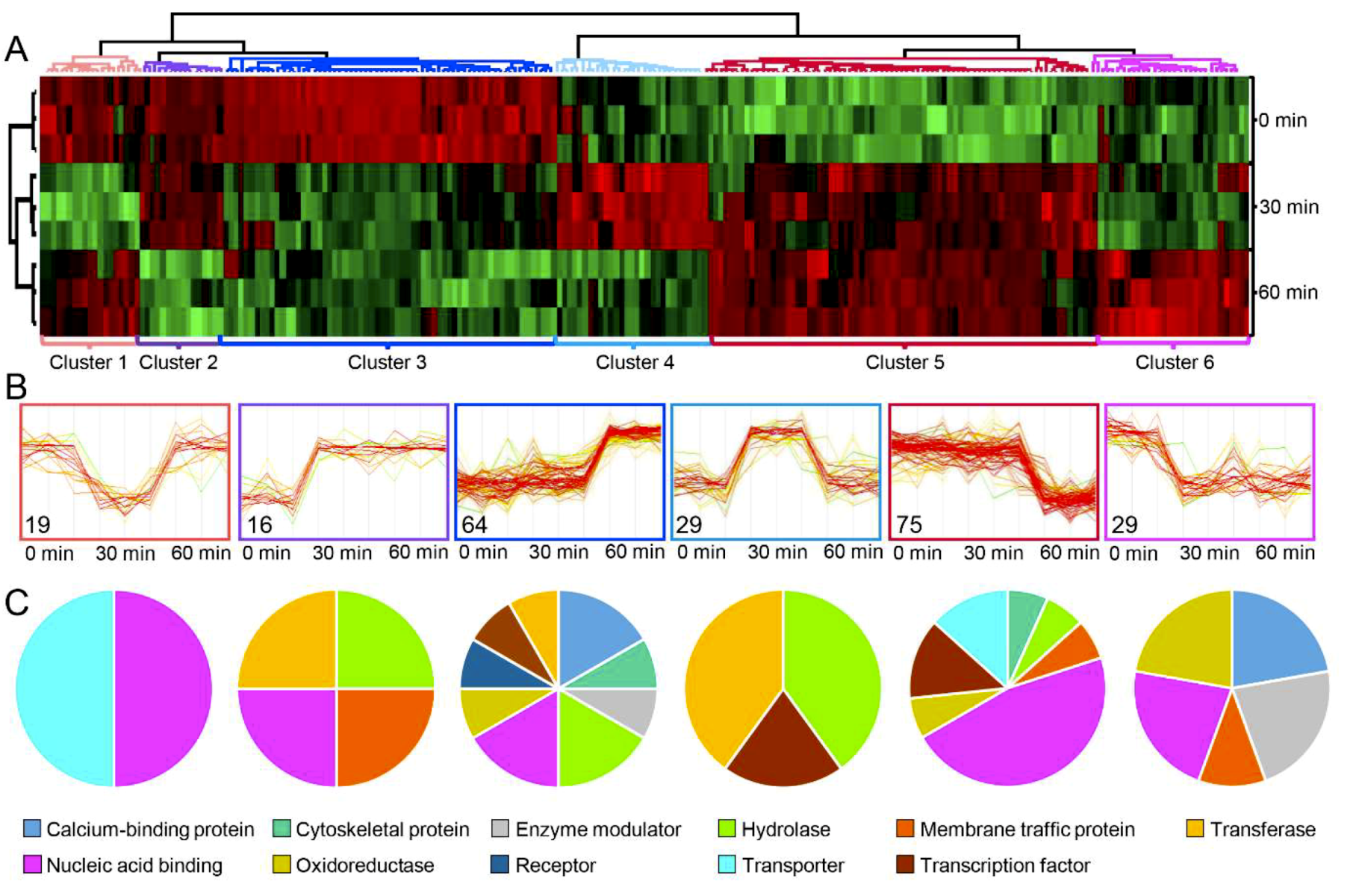
2.3. Phosphoproteome Analysis
3. Discussion
3.1. Activation of MAP Kinase Signaling by MSP1
3.2. MSP1 Treatment Leads to the Activation of ROS Detoxifying Enzymes
3.3. MSP1 Induced Signaling Is Mediated by Phytohormones
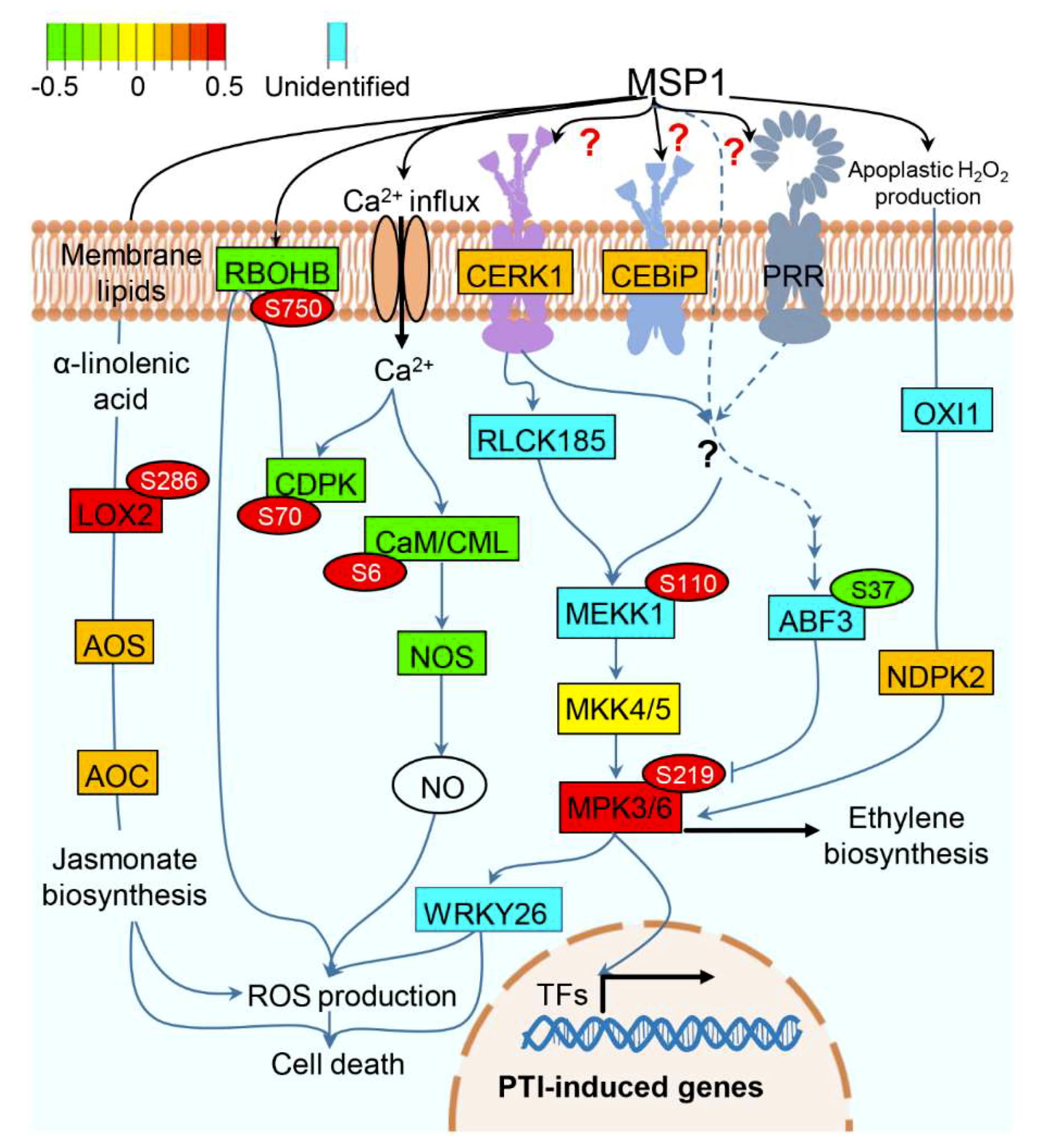
3.4. A Proposed Model to Elucidate the MSP1 Induced Signaling in Rice
4. Materials and Methods
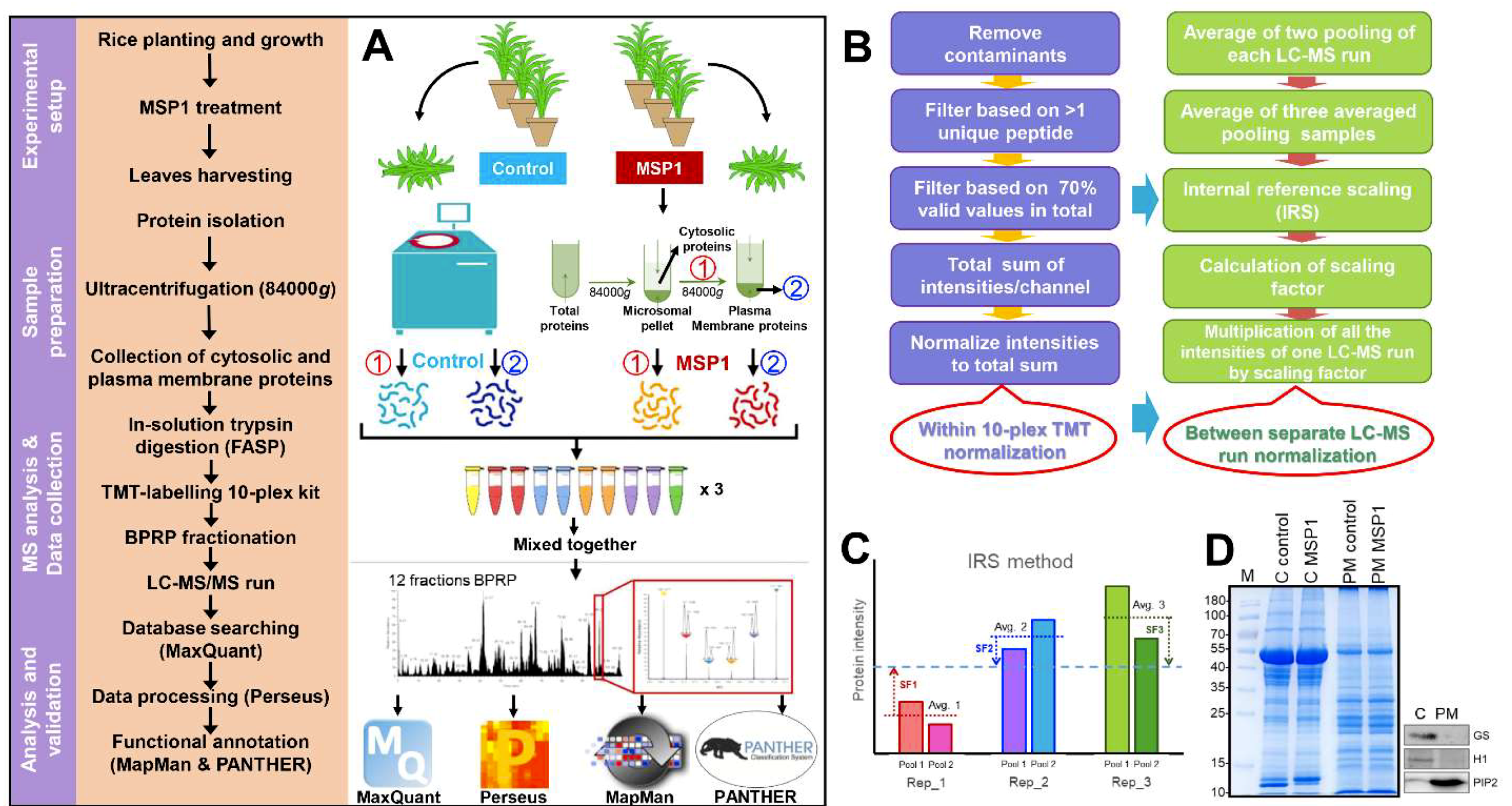
4.1. Plant Material, Growth Conditions, and Sample Preparation
4.2. Isolation of Cytosolic and PM-Localized Proteins
4.3. In-Solution Trypsin Digestion, Peptide Labeling, and Fractionation
4.4. Protein Extraction and Phosphopeptides Enrichment
4.5. Q-Exactive MS Analysis
4.6. LC-MS/MS Data Analysis for Proteomic Comparisons
4.7. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zipfel, C. Plant pattern-recognition receptors. Trends Immunol. 2014, 35, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lee, S.E.; Agrawal, G.K.; Rakwal, R.; Park, S.; Wang, Y.; Kim, S.T. Understanding the plant-pathogen interactions in the context of proteomics-generated apoplastic proteins inventory. Front. Plant Sci. 2015, 6, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Mitchell, T.K.; Dean, R.A. The Magnaporthe grisea snodprot1 homolog, MSP1, is required for virulence. FEMS Microbiol. Lett. 2007, 273, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zeng, H.; Qiu, D.; Guo, L.; Yang, X.; Shi, H.; Zhou, T.; Zhao, J. Purification and characterization of a novel hypersensitive response-inducing elicitor from Magnaporthe oryzae that triggers defense response in rice. PLoS ONE 2012, 7, e37654. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I. Cerato-platanin family proteins: One function for multiple biological roles? Front. Plant Sci. 2015, 5, 769. [Google Scholar] [CrossRef] [PubMed]
- Pazzagli, L.; Seidl-seiboth, V.; Barsottini, M.; Vargas, W.A.; Scala, A.; Mukherjee, P.K. Plant Science Cerato-platanins: Elicitors and effectors. Plant Sci. 2014, 228, 79–87. [Google Scholar] [CrossRef]
- Liu, S.; Wu, B.; Yang, J.; Bi, F.; Dong, T.; Yang, Q.; Hu, C.; Xiang, D.; Chen, H.; Huang, H. A Cerato-Platanin Family Protein FocCP1 Is Essential for the Penetration and Virulence of Fusarium oxysporum f. sp. cubense Tropical Race 4. Int. J. Mol. Sci. 2019, 20, 3785. [Google Scholar] [CrossRef]
- Gomes, E.V.; do Nascimento Costa, M.; De Paula, R.G.; De Azevedo, R.R.; Da Silva, F.L.; Noronha, E.F.; Ulhoa, C.J.; Monteiro, V.N.; Cardoza, R.E.; Gutiérrez, S. The Cerato-Platanin protein Epl-1 from Trichoderma harzianum is involved in mycoparasitism, plant resistance induction and self cell wall protection. Sci. Rep. 2015, 5, 17998. [Google Scholar] [CrossRef] [Green Version]
- Frías, M.; González, C.; Brito, N. BcSpl1, a cerato-platanin family protein, contributes to Botrytis cinerea virulence and elicits the hypersensitive response in the host. New Phytol. 2011, 192, 483–495. [Google Scholar] [CrossRef]
- Li, S.; Dong, Y.; Li, L.; Zhang, Y.; Yang, X.; Zeng, H.; Shi, M.; Pei, X.; Qiu, D.; Yuan, Q. The Novel Cerato-Platanin-Like Protein FocCP1 from Fusarium oxysporum Triggers an Immune Response in Plants. Int. J. Mol. Sci. 2019, 20, 2849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Kim, S.G.; Tsuda, K.; Gupta, R.; Park, S.-Y.; Kim, S.T.; Kang, K.Y. Magnaporthe oryzae-secreted protein MSP1 induces cell death and elicits defense responses in rice. Mol. Plant-Microbe Interact. 2016, 29, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Yang, Y.; Zhang, H.; Huang, L.; Li, D.; Song, F. Overexpression of MoSM1, encoding for an immunity-inducing protein from Magnaporthe oryzae, in rice confers broad-spectrum resistance against fungal and bacterial diseases. Sci. Rep. 2017, 7, 41037. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Gupta, R.; Min, C.W.; Kim, J.; Kramer, K.; Wang, Y.; Park, S.R.; Finkemeier, I.; Kim, S.T. A proteomic insight into the MSP1 and flg22 induced signaling in Oryza sativa leaves. J. Proteomics 2019, 196, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Park, J.; Han, D.; Yang, J.; Kim, A.; Woo, J.; Kim, Y.; Mook-Jung, I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 2018, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Plubell, D.L.; Wilmarth, P.A.; Zhao, Y.; Fenton, A.M.; Minnier, J.; Reddy, A.P.; Klimek, J.; Yang, X.; David, L.L.; Pamir, N. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteomics 2017, 16, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Quarantin, A.; Glasenapp, A.; Schäfer, W.; Favaron, F.; Sella, L. Involvement of the Fusarium graminearum cerato-platanin proteins in fungal growth and plant infection. Plant Physiol. Biochem. 2016, 109, 220–229. [Google Scholar] [CrossRef]
- Wilson, L.M.; Idnurm, A.; Howlett, B.J. Characterization of a gene (sp1) encoding a secreted protein from Leptosphaeria maculans, the blackleg pathogen of Brassica napus. Mol. Plant Pathol. 2002, 3, 487–493. [Google Scholar] [CrossRef]
- Djonović, S.; Pozo, M.J.; Dangott, L.J.; Howell, C.R.; Kenerley, C.M. Sm1, a proteinaceous elicitor secreted by the biocontrol fungus Trichoderma virens induces plant defense responses and systemic resistance. Mol. Plant-microbe Interact. 2006, 19, 838–853. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Liang, Y.; Dong, Y.; Yang, X.; Yuan, J.; Qiu, D. The Verticillium dahliae SnodProt1-like protein VdCP1 contributes to virulence and triggers the plant immune system. Front. Plant Sci. 2017, 8, 1880. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Gupta, R.; Min, C.W.; Kim, J.; Kramer, K.; Wang, Y.; Park, S.-R.; Finkemeier, I.; Kim, S.T. Label-free quantitative proteome data associated with MSP1 and flg22 induced signaling in rice leaves. Data Br. 2018, 20, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Coon, J.J. Phosphoproteomics in the age of rapid and deep proteome profiling. Anal. Chem. 2015, 88, 74–94. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Ni, L.; Liu, Y.; Zhang, A.; Tan, M.; Jiang, M. OsDMI3-mediated activation of OsMPK1 regulates the activities of antioxidant enzymes in abscisic acid signalling in rice. Plant. Cell Environ. 2014, 37, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Matsuoka, D.; Nanmori, T. Phosphorylation of Arabidopsisthaliana MEKK1 via Ca 2+ signaling as a part of the cold stress response. J. Plant Res. 2013, 126, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, F.; Jonak, C.; Ligterink, W.; Niehaus, K.; Boller, T.; Hirt, H. Differential activation of four specific MAPK pathways by distinct elicitors. J. Biol. Chem. 2000, 275, 36734–36740. [Google Scholar] [CrossRef] [PubMed]
- Devendrakumar, K.T.; Li, X.; Zhang, Y. MAP kinase signalling: Interplays between plant PAMP-and effector-triggered immunity. Cell. Mol. life Sci. 2018, 75, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Klessig, D.F. Salicylic Acid Activates a 48-kD MAP Kinase in Tobacco. Plant Cell 2007, 9, 809. [Google Scholar]
- Seo, S.; Sano, H.; Ohashi, Y. Jasmonate-based wound signal transduction requires activation of WIPK, a tobacco mitogen-activated protein kinase. Plant Cell 1999, 11, 289–298. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Kramer, K.; Agrawal, G.K.; Rakwal, R.; Park, K.; Wang, Y.; Finkemeier, I.; Kim, S.T. A Multi-Omics Analysis of Glycine max Leaves Reveals Alteration in Flavonoid and Isoflavonoid Metabolism upon Ethylene and Abscisic acid Treatment. Proteomics 2018. [Google Scholar] [CrossRef]
- Li, L.; Li, M.; Yu, L.; Zhou, Z.; Liang, X.; Liu, Z.; Cai, G.; Gao, L.; Zhang, X.; Wang, Y. The FLS2-associated kinase BIK1 directly phosphorylates the NADPH oxidase RbohD to control plant immunity. Cell Host Microbe 2014, 15, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Benschop, J.J.; Mohammed, S.; O’Flaherty, M.; Heck, A.J.R.; Slijper, M.; Menke, F.L.H. Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteomics 2007, 6, 1198–1214. [Google Scholar] [CrossRef] [PubMed]
- Nühse, T.S.; Bottrill, A.R.; Jones, A.M.E.; Peck, S.C. Quantitative phosphoproteomic analysis of plasma membrane proteins reveals regulatory mechanisms of plant innate immune responses. Plant, J. 2007, 51, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Seo, M.; Takebayashi, Y.; Kamiya, Y.; Riemann, M.; Nick, P. Jasmonates are induced by the PAMP flg22 but not the cell death-inducing elicitor Harpin in Vitis rupestris. Protoplasma 2017, 254, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Halim, V.A.; Altmann, S.; Ellinger, D.; Eschen-Lippold, L.; Miersch, O.; Scheel, D.; Rosahl, S. PAMP-induced defense responses in potato require both salicylic acid and jasmonic acid. Plant, J. 2009, 57, 230–242. [Google Scholar] [CrossRef]
- Kerk, D.; Bulgrien, J.; Smith, D.W.; Gribskov, M. Arabidopsis proteins containing similarity to the universal stress protein domain of bacteria. Plant Physiol. 2003, 131, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M. The novel ethylene-regulated gene OsUsp1 from rice encodes a member of a plant protein family related to prokaryotic universal stress proteins. J. Exp. Bot. 2002, 53, 2325–2331. [Google Scholar] [CrossRef]
- Gutiérrez-Beltrán, E.; Personat, J.M.; de la Torre, F.; del Pozo, O. A universal stress protein involved in oxidative stress is a phosphorylation target for protein kinase CIPK6. Plant Physiol. 2017, 173, 836–852. [Google Scholar] [CrossRef]
- Merkouropoulos, G.; Andreasson, E.; Hess, D.; Boller, T.; Peck, S.C. An Arabidopsis protein phosphorylated in response to microbial elicitation, AtPHOS32, is a substrate of MAP kinases 3 and 6. J. Biol. Chem. 2008, 283, 10493–10499. [Google Scholar] [CrossRef]
- Mine, A.; Berens, M.L.; Nobori, T.; Anver, S.; Fukumoto, K.; Winkelmüller, T.M.; Takeda, A.; Becker, D.; Tsuda, K. Pathogen exploitation of an abscisic acid-and jasmonate-inducible MAPK phosphatase and its interception by Arabidopsis immunity. Proc. Natl. Acad. Sci. USA 2017, 114, 7456–7461. [Google Scholar] [CrossRef]
- Reinbothe, C.; Springer, A.; Samol, I.; Reinbothe, S. Plant oxylipins: Role of jasmonic acid during programmed cell death, defence and leaf senescence. FEBS J. 2009, 276, 4666–4681. [Google Scholar] [CrossRef]
- Santoni, V. Plant plasma membrane protein extraction and solubilization for proteomic analysis. In Plant Proteomics; Springer: Totowa, NJ, USA, 2007; pp. 93–109. [Google Scholar]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Meng, Q.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Comparative phosphoproteome analysis upon ethylene and abscisic acid treatment in Glycine max leaves. Plant Physiol. Biochem. 2018, 130, 173–180. [Google Scholar] [CrossRef]
- Gupta, R.; Lee, S.J.; Min, C.W.; Kim, S.W.; Park, K.-H.; Bae, D.-W.; Lee, B.W.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Coupling of gel-based 2-DE and 1-DE shotgun proteomics approaches to dig deep into the leaf senescence proteome of Glycine max. J. Proteomics 2016, 148, 65–74. [Google Scholar] [CrossRef]
- Min, C.W.; Lee, S.H.; Cheon, Y.E.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Gupta, R.; et al. In-depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. J. Proteomics 2017, 169, 125–135. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]
- Mann, M. MaxQuant enables high peptide identification rates. Online 2008, 26, 1–6. [Google Scholar]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Meng, Q.; Jun, T.H.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Phosphoproteome data from abscisic acid and ethylene treated Glycine max leaves. Data Br. 2018, 20, 516–520. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Snel, B. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TMT plex & technical replicates | Sample Labeling | Number of identified proteins | Total number of identified peptides | Average Sequence Coverage/protein | |||||
|---|---|---|---|---|---|---|---|---|---|
| TMT Reagent | Sample Info | Cytosolic | PM | Cytosolic | PM | Cytosolic | PM | ||
| TMT plex set 1 | Technical Replicate 1 | 126 | Control_1 | 3329 | 3405 | 23,680 | 22,287 | 21.06 | 19.24 |
| 127N | MSP1_1 | 3342 | 3397 | ||||||
| 127C | Control_2 | 3349 | 3416 | ||||||
| 128N | MSP1_2 | 3349 | 3410 | ||||||
| 128C | Control_3 | 3340 | 3418 | ||||||
| 129N | MSP1_3 | 3336 | 3419 | ||||||
| 129C | Control_4 | 3338 | 3424 | ||||||
| 130N | MSP1_4 | 3339 | 3416 | ||||||
| 130C | Pooling 1 | 3344 | 3421 | ||||||
| 131 | Pooling 2 | 3340 | 3413 | ||||||
| TMT plex set 2 | Technical Replicate 2 | 126 | Control_1 | 3257 | 3432 | 23,672 | 22,324 | 21.1 | 19.32 |
| 127N | MSP1_1 | 3281 | 3433 | ||||||
| 127C | Control_2 | 3273 | 3448 | ||||||
| 128N | MSP1_2 | 3271 | 3430 | ||||||
| 128C | Control_3 | 3274 | 3458 | ||||||
| 129N | MSP1_3 | 3286 | 3445 | ||||||
| 129C | Control_4 | 3271 | 3450 | ||||||
| 130N | MSP1_4 | 3275 | 3439 | ||||||
| 130C | Pooling 1 | 3289 | 3449 | ||||||
| 131 | Pooling 2 | 3270 | 3442 | ||||||
| TMT plex set 3 | Technical Replicate 3 | 126 | Control_1 | 3326 | 3423 | 23,841 | 22,358 | 21.28 | 19.34 |
| 127N | MSP1_1 | 3348 | 3420 | ||||||
| 127C | Control_2 | 3331 | 3426 | ||||||
| 128N | MSP1_2 | 3344 | 3419 | ||||||
| 128C | Control_3 | 3332 | 3433 | ||||||
| 129N | MSP1_3 | 3335 | 3429 | ||||||
| 129C | Control_4 | 3321 | 3444 | ||||||
| 130N | MSP1_4 | 3335 | 3429 | ||||||
| 130C | Pooling 1 | 3340 | 3421 | ||||||
| 131 | Pooling 2 | 3338 | 3431 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, R.; Min, C.W.; Kim, Y.-J.; Kim, S.T. Identification of Msp1-Induced Signaling Components in Rice Leaves by Integrated Proteomic and Phosphoproteomic Analysis. Int. J. Mol. Sci. 2019, 20, 4135. https://doi.org/10.3390/ijms20174135
Gupta R, Min CW, Kim Y-J, Kim ST. Identification of Msp1-Induced Signaling Components in Rice Leaves by Integrated Proteomic and Phosphoproteomic Analysis. International Journal of Molecular Sciences. 2019; 20(17):4135. https://doi.org/10.3390/ijms20174135
Chicago/Turabian StyleGupta, Ravi, Cheol Woo Min, Yu-Jin Kim, and Sun Tae Kim. 2019. "Identification of Msp1-Induced Signaling Components in Rice Leaves by Integrated Proteomic and Phosphoproteomic Analysis" International Journal of Molecular Sciences 20, no. 17: 4135. https://doi.org/10.3390/ijms20174135
APA StyleGupta, R., Min, C. W., Kim, Y. -J., & Kim, S. T. (2019). Identification of Msp1-Induced Signaling Components in Rice Leaves by Integrated Proteomic and Phosphoproteomic Analysis. International Journal of Molecular Sciences, 20(17), 4135. https://doi.org/10.3390/ijms20174135