New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment

Abstract
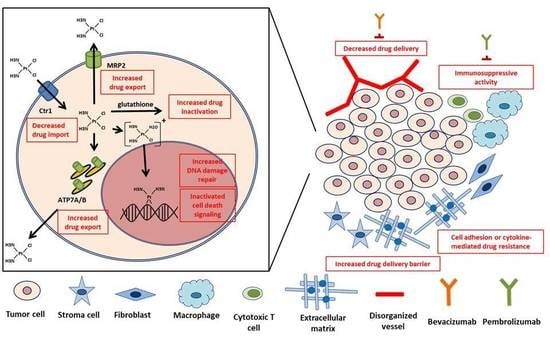
:
1. Introduction
2. Mechanism of CDDP-Induced Cytotoxicity
3. Conventional Perspectives on CDDP Resistance from a Tumor Cell
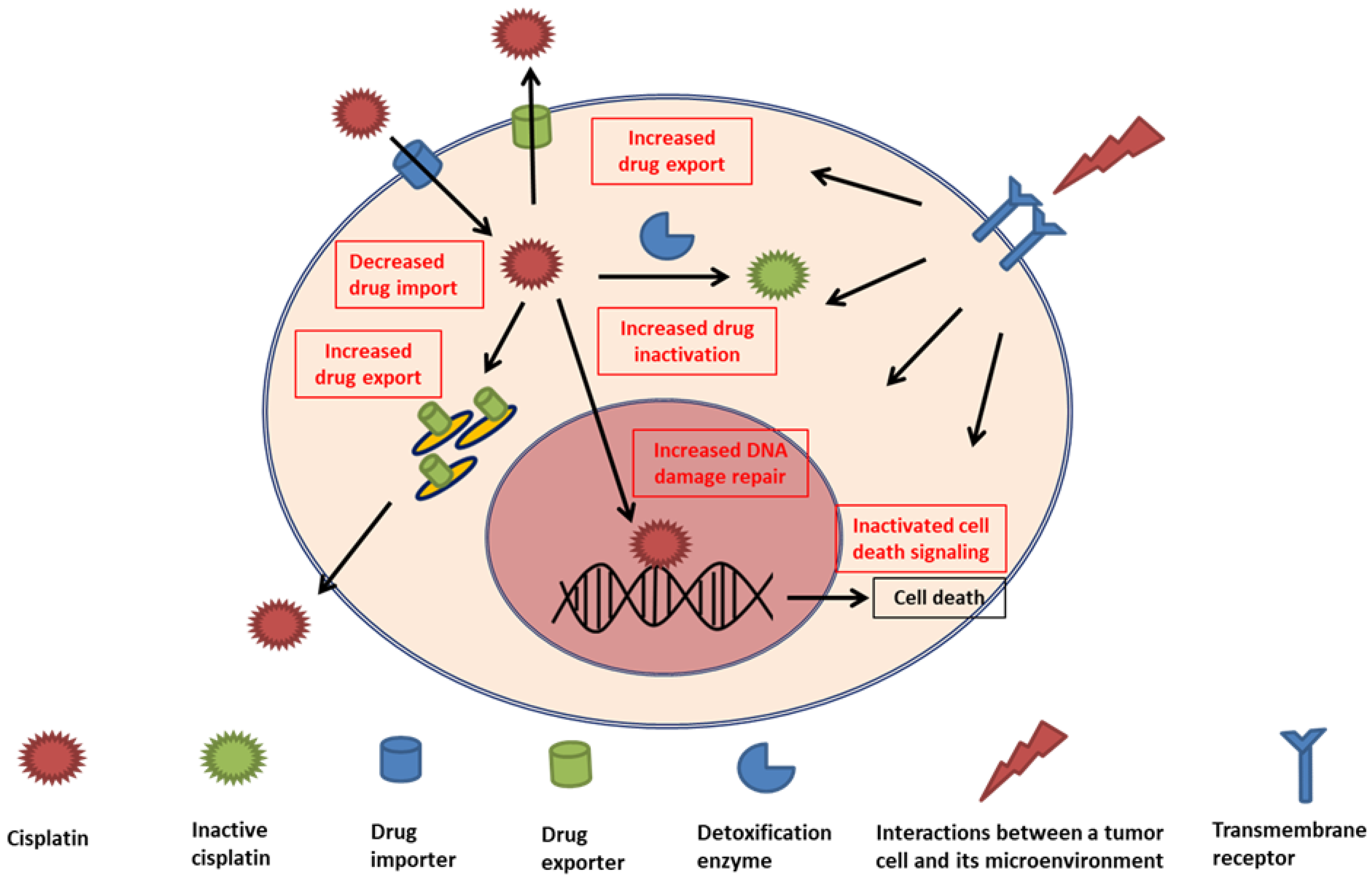
3.1. Cellular Accumulation of Drug
3.1.1. Decrease in Uptake
3.1.2. Increase in Efflux
3.2. Intracellular Drug Detoxification
3.3. DNA Damage Repair
3.3.1. Nucleotide Excision Repair
3.3.2. BRCA1/BRCA2
3.3.3. Other DNA Repair Participants
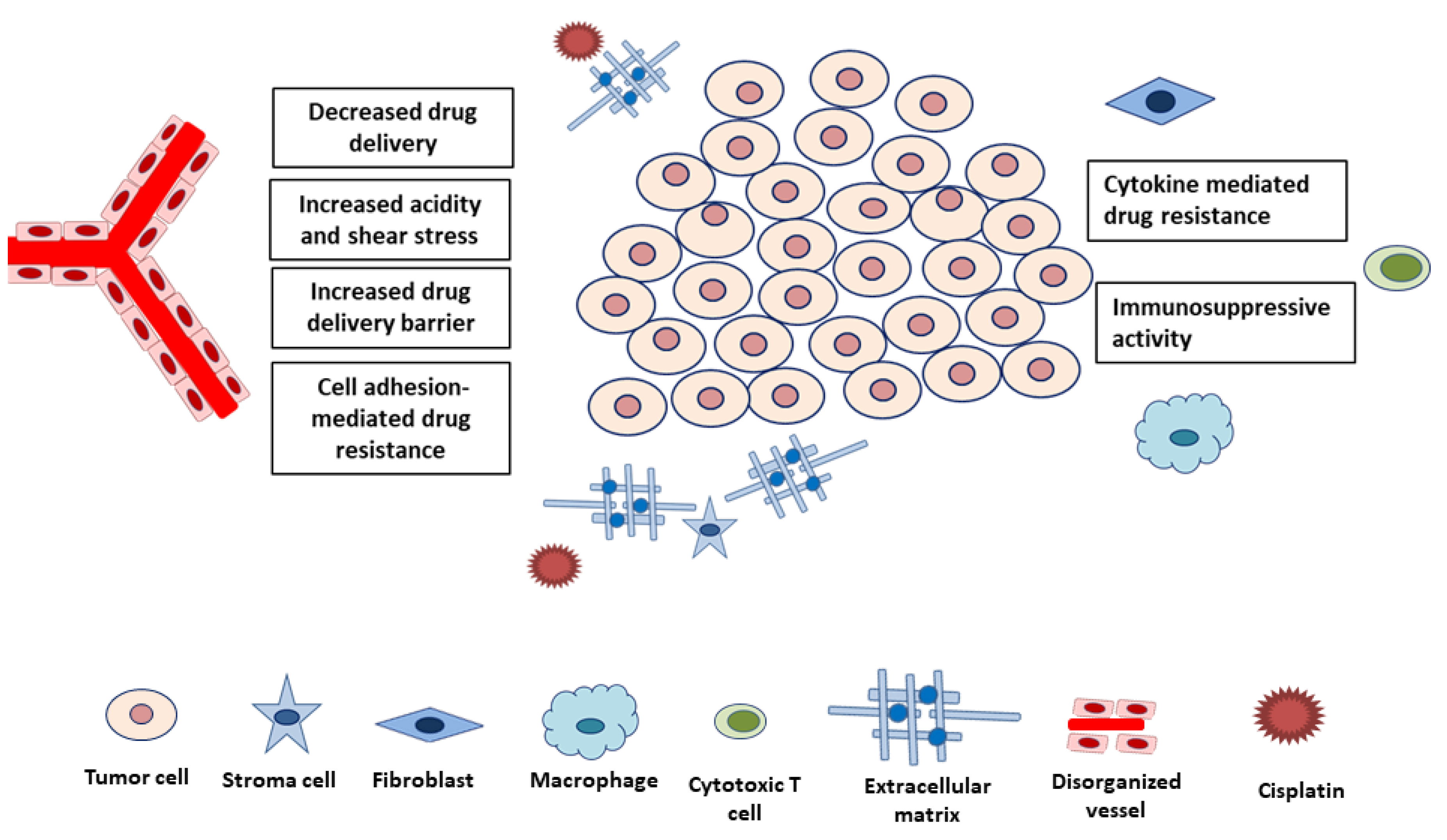
4. Emerging Perspectives on CDDP Resistance from the TME
4.1. Physical Factors
4.2. Biological Factors
4.2.1. Reduced Blood Flow
4.2.2. Cellular Crosstalk within The Microenvironment
4.2.3. Immune System
5. Ongoing Approaches to Overcome CDDP Resistance
6. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 5, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum antitumor complexes: 50 years since Barnett Rosenberg’s discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg Chem. 2019, 11, 88:102925. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat. 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.; Choi, N.; Kim, K.; Koo, H.J.; Choi, J.; Kim, H.N. Chemoresistance of cancer cells: Requirements of tumor microenvironment-mimicking in vitro models in anti-cancer drug development. Theranostics 2018, 8, 5259–5275. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, W.C.; Liang, Z.D.; Tsai, W.B.; Long, Y.; Aiba, I.; Fu, S.; Broaddus, R.; Liu, J.; Feun, L.G.; et al. Targeting drug transport mechanisms for improving platinum-based cancer chemotherapy. Expert Opin. Ther. Targets. 2015, 19, 1307–1317. [Google Scholar] [CrossRef]
- Davies, M.S.; Berners-Price, S.J.; Hambley, T.W. Slowing of cisplatin aquation in the presence of DNA but not in the presence of phosphate: Improved understanding of sequence selectivity and the roles of monoaquated and diaquated species in the binding of cisplatin to DNA. Inorg. Chem. 2000, 39, 5603–5613. [Google Scholar] [CrossRef]
- Eastman, A. Cross-linking of glutathione to DNA by cancer chemotherapeutic platinum coordination complexes. Chem. Biol. Interact. 1987, 61, 241–248. [Google Scholar] [CrossRef]
- El-Khateeb, M.; Appleton, T.G.; Gahan, L.R. Charles BG, Berners-Price SJ, Bolton AM. Reactions of cisplatin hydrolytes with methionine, cysteine, and plasma ultrafiltrate studied by a combination of HPLC and NMR techniques. J. Inorg. Biochem. 1999, 77, 13–21. [Google Scholar] [CrossRef]
- Eastman, A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol. Ther. 1987, 34, 155–166. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B. Menck CFM. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73. [Google Scholar] [CrossRef]
- Rebillard, A.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.T. Cisplatin cytotoxicity: DNA and plasma membrane targets. Curr. Med. Chem. 2008, 15, 2656–2663. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; Prieto-García, L.; Prieto, M.; López-Novoa, J.M.; López-Hernández, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef]
- Chen, S.J.; Kuo, C.C.; Pan, H.Y.; Tsou, T.C.; Yeh, S.C.; Chang, J.Y. Mechanistic basis of a combination D-penicillamine and platinum drugs synergistically inhibits tumor growth in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo. Biochem. Pharmacol. 2015, 95, 28–37. [Google Scholar] [CrossRef]
- Chen, S.J.; Kuo, C.C.; Pan, H.Y.; Tsou, T.C.; Yeh, S.C.; Chang, J.Y. Desferal regulates hCtr1 and transferrin receptor expression through Sp1 and exhibits synergistic cytotoxicity with platinum drugs in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo. Oncotarget 2016, 7, 49310–49321. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar]
- Kim, E.S.; Tang, X.; Peterson, D.R.; Kilari, D.; Chow, C.W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Stewart, D.J.; Wistuba, I.I.; et al. Copper transporter CTR1 expression and tissue platinum concentration in non-small cell lung cancer. Lung Cancer 2014, 85, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010, 17, 574–583. [Google Scholar] [CrossRef]
- Yang, T.; Chen, M.; Chen, T.; Thakur, A. Expression of the copper transporters hCtr1.; ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 2015, 10, 2584–2590. [Google Scholar] [CrossRef]
- Öhrvik, H.; Logeman, B.; Turk, B.; Reinheckel, T.; Thiele, D.J. Cathepsin protease controls copper and cisplatin accumulation via cleavage of the Ctr1 metal-binding ectodomain. J. Biol. Chem. 2016, 291, 13905–13916. [Google Scholar]
- Lee, Y.Y.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, W.; Park, H.S.; Song, T.J.; Kim, M.K.; Kim, T.J.; Lee, J.W.; et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy. Gynecol. Oncol. 2011, 122, 361–365. [Google Scholar] [CrossRef]
- Yoshida, H.; Teramae, M.; Yamauchi, M.; Fukuda, T.; Yasui, T.; Sumi, T.; Honda, K.; Ishiko, O. Association of copper transporter expression with platinum resistance in epithelial ovarian cancer. Anticancer Res. 2013, 33, 1409–1414. [Google Scholar]
- Naka, A.; Takeda, R.; Shintani, M.; Ogane, N.; Kameda, Y.; Aoyama, T.; Yoshikawa, T.; Kamoshida, S. Organic cation transporter 2 for predicting cisplatin-based neoadjuvant chemotherapy response in gastric cancer. Am. J. Cancer Res. 2015, 5, 2285–2293. [Google Scholar]
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin Cancer Res. 2003, 9, 5853–5859. [Google Scholar]
- Miyashita, H.; Nitta, Y.; Mori, S.; Kanzaki, A.; Nakayama, K.; Terada, K.; Sugiyama, T.; Kawamura, H.; Sato, A.; Morikawa, H.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a chemoresistance marker in human oral squamous cell carcinoma treated with cisplatin. Oral Oncol. 2003, 39, 157–162. [Google Scholar] [CrossRef]
- Higashimoto, M.; Kanzaki, A.; Shimakawa, T.; Konno, S.; Naritaka, Y.; Nitta, Y.; Mori, S.; Shirata, S.; Yoshida, A.; Terada, K.; et al. Expression of copper-transporting P-type adenosine triphosphatase in human esophageal carcinoma. Int. J. Mol Med. 2003, 11, 337–341. [Google Scholar] [CrossRef]
- Nakayama, K.; Kanzaki, A.; Terada, K.; Mutoh, M.; Ogawa, K.; Sugiyama, T.; Takenoshita, S.; Itoh, K.; Yaegashi, N.; Miyazaki, K.; et al. Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin. Cancer Res. 2004, 10, 2804–2811. [Google Scholar] [CrossRef]
- Hinoshita, E.; Uchiumi, T.; Taguchi, K.; Kinukawa, N.; Tsuneyoshi, M.; Maehara, Y.; Sugimachi, K.; Kuwano, M. Increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomas. Clin. Cancer Res. 2000, 6, 2401–2407. [Google Scholar]
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K.; et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972. [Google Scholar]
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer. 2011, 104, 707–713. [Google Scholar] [CrossRef]
- Lewis, A.D.; Hayes, J.D.; Wolf, C.R. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: Intrinsic differences and cell cycle effects. Carcinogenesis 1988, 9, 1283–1287. [Google Scholar] [CrossRef]
- Hirano, T.; Kato, H.; Maeda, M.; Gong, Y.; Shou, Y.; Nakamura, M.; Maeda, J.; Yashima, K.; Kato, Y.; Akimoto, S.; et al. Identification of postoperative adjuvant chemotherapy responders in non-small cell lung cancer by novel biomarker. Int. J. Cancer. 2005, 117, 460–468. [Google Scholar] [CrossRef]
- Surowiak, P.; Materna, V.; Kaplenko, I.; Spaczyński, M.; Dietel, M.; Lage, H.; Zabel, M. Augmented expression of metallothionein and glutathione S-transferase pi as unfavourable prognostic factors in cisplatin-treated ovarian cancer patients. Virchows. Arch. 2005, 447, 626–633. [Google Scholar] [CrossRef]
- Kasahara, K.; Fujiwara, Y.; Nishio, K.; Ohmori, T.; Sugimoto, Y.; Komiya, K.; Matsuda, T.; Saijo, N. Metallothionein content correlates with the sensitivity of human small cell lung cancer cell lines to cisplatin. Cancer Res. 1991, 51, 3237–3242. [Google Scholar]
- Hishikawa, Y.; Abe, S.; Kinugasa, S.; Yoshimura, H.; Monden, N.; Igarashi, M.; Tachibana, M.; Nagasue, N. Overexpression of metallothionein correlates with chemoresistance to cisplatin and prognosis in esophageal cancer. Oncology 1997, 54, 342–347. [Google Scholar] [CrossRef]
- Deloia, J.A.; Bhagwat, N.R.; Darcy, K.M.; Strange, M.; Tian, C.; Nuttall, K.; Krivak, T.C.; Niedernhofer, L.J. Comparison of ERCC1/XPF genetic variation, mRNA and protein levels in women with advanced stage ovarian cancer treated with intraperitoneal platinum. Gynecol. Oncol. 2012, 126, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Qing, Y.; Guan, W.; Li, M.; Peng, Y.; Zhang, S.; Xiong, Y.; Wang, D. Predictive value of APE1, BRCA1, ERCC1 and TUBB3 expression in patients with advanced non-small cell lung cancer (NSCLC) receiving first-line platinum-paclitaxel chemotherapy. Cancer Chemother. Pharmacol. 2014, 74, 777–786. [Google Scholar] [CrossRef]
- Villalobos, M.; Czapiewski, P.; Reinmuth, N.; Fischer, J.R.; Andreas, S.; Kortsik, C.; Serke, M.; Wolf, M.; Neuser, P.; Reuss, A.; et al. ERCC1 assessment in upfront treatment with and without cisplatin-based chemotherapy in stage IIIB/IV non-squamous non-small cell lung cancer. Med. Oncol. 2018, 35, 106. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, C.; Hu, L. Low expression of excision repair cross-complementation group-1 protein predicts better outcome in patients with locally advanced nasopharyngeal cancer treated with concurrent chemoradiotherapy. Tumori 2014, 100, 328–332. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, Y.; Zhang, H.; Qu, T.; Mao, Y.; Zhu, H.; Quan, L.; Xing, P.; Wang, J.; He, J.; et al. A phase II study of biweekly paclitaxel and cisplatin chemotherapy for recurrent or metastatic esophageal squamous cell carcinoma: ERCC1 expression predicts response to chemotherapy. Med. Oncol. 2013, 30, 343. [Google Scholar] [CrossRef]
- Ryu, H.; Song, I.C.; Choi, Y.S.; Yun, H.J.; Jo, D.Y.; Kim, J.M.; Ko, Y.B.; Lee, H.J. ERCC1 expression status predicts the response and survival of patients with metastatic or recurrent cervical cancer treated via platinum-based chemotherapy. Medicine 2017, 96, e9402. [Google Scholar] [CrossRef]
- Xuelei, M.; Jingwen, H.; Wei, D.; Hongyu, Z.; Jing, Z.; Changle, S.; Lei, L. ERCC1 plays an important role in predicting survival outcomes and treatment response for patients with HNSCC: A meta-analysis. Oral Oncol. 2015, 51, 483–492. [Google Scholar] [CrossRef]
- Hwang, I.G.; Jang, J.S.; Do, J.H.; Kang, J.H.; Lee, G.W.; Oh, S.Y.; Kwon, H.C.; Jun, H.J.; Lim, H.Y.; Lee, S.; et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 2011, 8, 935–944. [Google Scholar] [CrossRef]
- Vaezi, A.; Wang, X.; Buch, S.; Gooding, W.; Wang, L.; Seethala, R.R.; Weaver, D.T.; D’Andrea, A.D.; Argiris, A.; Romkes, M.; et al. XPF expression correlates with clinical outcome in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2011, 17, 5513–5522. [Google Scholar] [CrossRef]
- Gourley, C.; Michie, C.O.; Roxburgh, P.; Yap, T.A.; Harden, S.; Paul, J.; Ragupathy, K.; Todd, R.; Petty, R.; Reed, N.; et al. Increased incidence of visceral metastases in scottish patients with BRCA1/2-defective ovarian cancer: An extension of the ovarian BRCAness phenotype. J. Clin. Oncol. 2010, 28, 2505–2511. [Google Scholar] [CrossRef]
- Lowery, M.A.; Kelsen, D.P.; Stadler, Z.K.; Yu, K.H.; Janjigian, Y.Y.; Ludwig, E.; D’Adamo, D.R.; Salo-Mullen, E.; Robson, M.E.; Allen, P.J.; et al. An emerging entity: Pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011, 16, 1397–1402. [Google Scholar] [CrossRef]
- Chen, S.H.; Kuo, C.C.; Li, C.F.; Cheung, C.H.; Tsou, T.C.; Chiang, H.C.; Yang, Y.N.; Chang, S.L.; Lin, L.C.; Pan, H.Y.; et al. O(6) -methylguanine DNA methyltransferase repairs platinum-DNA adducts following cisplatin treatment and predicts prognoses of nasopharyngeal carcinoma. Int. J. Cancer 2015, 137, 1291–1305. [Google Scholar] [CrossRef]
- Lai, Y.H.; Kuo, C.; Kuo, M.T.; Chen, H.H.W. Modulating chemosensitivity of tumors to platinum-based antitumor drugs by transcriptional regulation of copper homeostasis. Int. J. Mol Sci. 2018, 19, 1486. [Google Scholar] [CrossRef]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the human organic cation transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS. Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes. Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. 2014, 32, 806–814. [Google Scholar] [CrossRef]
- Cescon, D.W.; She, D.; Sakashita, S.; Zhu, C.Q.; Pintilie, M.; Shepherd, F.A.; Tsao, M.S. NRF2 pathway activation and adjuvant chemotherapy benefit in lung squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 2499–2505. [Google Scholar] [CrossRef]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer. 2012, 12, 587–598. [Google Scholar] [CrossRef]
- The NCCN Clinical Practice Guidelines in Oncology Home Page. Available online: https://www.nccn.org (accessed on 16 June 2019).
- Zhang, J.; Zhu, Y.; Wang, Y.; Fu, Q.; Xie, H.; Liu, Z.; Fu, H.; Cao, Y.; Xu, J.; Dai, B. Prognostic and predictive value of O6-methylguanine methyltransferase for chemotherapy in patients with muscle-invasive bladder cancer. Ann. Surg. Oncol. 2018, 25, 342–348. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.; Christmann, M. Targeting O6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-induced antitumor immunomodulation: A review of preclinical and clinical evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Ip, C.K.; Li, S.S.; Tang, M.Y.; Sy, S.K.; Ren, Y.; Shum, H.C.; Wong, A.S. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci. Rep. 2016, 6, 26788. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int. J. Mol Sci. 2018, 19. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Zhao, W.; Xia, S.Q.; Zhuang, J.P.; Zhang, Z.P.; You, C.C.; Yan, J.L.; Xu, G.P. Hypoxia-induced resistance to cisplatin-mediated apoptosis in osteosarcoma cells is reversed by gambogic acid independently of HIF-1α. Mol. Cell. Biochem. 2016, 420, 1–8. [Google Scholar] [CrossRef]
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. A drug combination targeting hypoxia induced chemoresistance and stemness in glioma cells. Oncotarget. 2018, 9, 18351–18366. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.C.; Hwang, S.H.; Kim, N.Y.; Lee, H.S.; Ji, S.; Yang, Y.; Kim, Y. Hypoxia promotes acquisition of aggressive phenotypes in human malignant mesothelioma. BMC. Cancer. 2018, 18, 819. [Google Scholar] [CrossRef]
- Soleymani Abyaneh, H.; Gupta, N.; Alshareef, A.; Gopal, K.; Lavasanifar, A.; Lai, R. Hypoxia induces the acquisition of cancer stem-like phenotype via upregulation and activation of signal transducer and activator of rranscription-3 (STAT3) in MDA-MB-231, a triple negative breast cancer cell line. Cancer Microenviron. 2018, 11, 141–152. [Google Scholar] [CrossRef]
- Raghunand, N.; Gillies, R.J. pH and drug resistance in tumors. Drug Resist. Update. 2000, 3, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Gerweck, L.E.; Vijayappa, S.; Kozin, S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol. Cancer Ther. 2006, 5, 1275–1279. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene. 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620. [Google Scholar] [CrossRef]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Salvagno, C.; Ciampricotti, M.; Tuit, S.; Hau, C.S.; van Weverwijk, A.; Coffelt, S.B.; Kersten, K.; Vrijland, K.; Kos, K.; Ulas, T.; et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat. Cell Biol. 2019, 21, 511–521. [Google Scholar] [CrossRef]
- Pass, H.I.; Lavilla, C.; Canino, C.; Goparaju, C.; Preiss, J.; Noreen, S.; Blandino, G.; Cioce, M. Inhibition of the colony-stimulating-factor-1 receptor affects the resistance of lung cancer cells to cisplatin. Oncotarget 2016, 7, 56408–56421. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Sato, N.; Kohi, S.; Hirata, K.; Goggins, M. Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. 2016, 107, 569–575. [Google Scholar] [CrossRef]
- Wong, P.P.; Bodrug, N.; Hodivala-Dilke, K.M. Exploring novel methods for modulating tumor blood vessels in cancer treatment. Curr. Biol. 2016, 26, 1161–1166. [Google Scholar] [CrossRef]
- Reck, M.; von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova, V.; Hirsh, V.; Leighl, N.; Mezger, J.; Archer, V.; Moore, N.; et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil. J. Clin. Oncol. 2009, 27, 1227–1234. [Google Scholar] [CrossRef]
- Barlesi, F.; Scherpereel, A.; Rittmeyer, A.; Pazzola, A.; Ferrer Tur, N.; Kim, J.H.; Ahn, M.J.; Aerts, J.G.; Gorbunova, V.; Vikström, A.; et al. Randomized phase III trial of maintenance bevacizumab with or without pemetrexed after first-line induction with bevacizumab, cisplatin, and pemetrexed in advanced nonsquamous non-small-cell lung cancer: AVAPERL (MO22089). J. Clin. Oncol. 2013, 31, 3004–3011. [Google Scholar] [CrossRef]
- Castells, M.; Thibault, B.; Delord, J.P.; Couderc, B. Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [Green Version]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Samuel, P.; Mulcahy, L.A.; Furlong, F.; McCarthy, H.O.; Brooks, S.A.; Fabbri, M.; Pink, R.C. Carter DRF. Cisplatin induces the release of extracellular vesicles from ovarian cancer cells that can induce invasiveness and drug resistance in bystander cells. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2018, 373, 1737. [Google Scholar] [CrossRef]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of RAB7A protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52. [Google Scholar] [CrossRef]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 12. [Google Scholar] [CrossRef]
- Jackaman, C.; Majewski, D.; Fox, S.A.; Nowak, A.K.; Nelson, D.J. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol. Immunother. 2012, 61, 2343–2356. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. 2015, 21, 5047–5056. [Google Scholar]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef]
- Wang, W.; Kryczek, I.; Dostál, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef]
- Beyranvand Nejad, E.; van der Sluis, T.C.; van Duikeren, S.; Yagita, H.; Janssen, G.M.; van Veelen, P.A.; Melief, C.J.; van der Burg, S.H.; Arens, R. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Cancer Res. 2016, 76, 6017–6029. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. IMpower150 Study Group. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Yeh, Y.M.; Hsu, S.J.; Lin, P.C.; Hsu, K.F.; Wu, P.Y.; Su, W.C.; Chang, J.Y.; Shen, M.R. The c.1085A>G genetic variant of CSF1R gene regulates tumor immunity by altering the proliferation, polarization, and function of macrophages. Clin. Cancer Res. 2017, 23, 6021–6030. [Google Scholar] [CrossRef]
- Fu, S.; Hou, M.M.; Wheler, J.; Hong, D.; Naing, A.; Tsimberidou, A.; Janku, F.; Zinner, R.; Piha-Paul, S.; Falchook, G.; et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Invest. New Drugs. 2014, 32, 465–472. [Google Scholar] [CrossRef]
- Dancey, J.E.; Chen, H.X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 649. [Google Scholar] [CrossRef]
- Chen, X.J.; Zhang, X.Q.; Liu, Q.; Zhang, J.; Zhou, G. Nanotechnology: A promising method for oral cancer detection and diagnosis. J. Nanobiotechnol. 2018, 16, 52. [Google Scholar] [CrossRef]
- Boulikas, T. Low toxicity and anticancer activity of a novel liposomal cisplatin (Lipoplatin) in mouse xenografts. Oncol Rep. 2004, 12, 3–12. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Gervais, A.C.; Egorin, M.J.; Schellens, J.H.; Zuhowski, E.G.; Pluim, D.; Joseph, E.; Hamburger, D.R.; Working, P.K.; et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother. Pharmacol. 2004, 53, 329–336. [Google Scholar] [CrossRef]
- Kudo, M.; Yamamoto, Y.; Koga, Y.; Hamaguchi, T.; Akimoto, T.; Yasunaga, M.; Matsumura, Y. Effect of combined treatment with micelle-incorporated cisplatin (NC-6004) and S-1 on human gastric cancer xenografts. Mol. Clin. Oncol. 2016, 5, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Cabral, H.; Nishiyama, N.; Okazaki, S.; Koyama, H.; Kataoka, K. Preparation and biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-loaded polymeric micelles. J. Control Release. 2005, 101, 223–232. [Google Scholar] [CrossRef]
- Farooq, M.A.; Aquib, M.; Farooq, A.; Haleem Khan, D.; Joelle Maviah, M.B.; Sied Filli, M.; Kesse, S.; Boakye-Yiadom, K.O.; Mavlyanova, R.; Parveen, A.; et al. Recent progress in nanotechnology-based novel drug delivery systems in designing of cisplatin for cancer therapy: An overview. Artif. Cells Nanomed Biotechnol. 2019, 47, 1674–1692. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Regulator | Action Mechanism | Relevance to CDDP Resistance | Reference |
|---|---|---|---|
| Cellular uptake | |||
| CTR1 | Membrane copper transporter | 1. Low expression levels in CDDP-resistant cancer cells. 2. Correlation between CTR1 expression levels and intracellular platinum concentration. 3. Copper chelators enhance CDDP efficacy in vitro and in vivo. 4. Low expression levels in tumors predict poor clinical efficacy of CDDP. | [16,17,18,19,20,21] |
| CTR2 | Membrane copper transporter | 1. The induction of CTR1 cleavage. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [22,23,24] |
| OCT2 | Organic cation transporter | Low expression levels in tumors predict poor clinical efficacy of CDDP. | [25] |
| Cellular export | |||
| ATP7A/ATP7B | Copper-exporting P-type ATPase | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [21,26,27,28,29] |
| MRP2 | ATP-binding cassette multidrug transporter | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [30,31,32] |
| Drug inactivation | |||
| GSH | Intracellular electrophiles scavenger | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [33,34,35] |
| Metallothionein | Detoxification enzyme of a heavy metal | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [36,37] |
| DNA damage repair | |||
| ERCC1 | NER | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [38,39,40,41,42,43,44,45] |
| XPF | NER | 1. High expression levels in CDDP-resistant cancer cells. 2. High expression levels in tumors predict poor clinical efficacy of CDDP. | [46] |
| BRCA1/BRCA2 | HR | BRCA1/2-mutated tumors correlate to good responders to CDDP. | [47,48] |
| Factor | Action mechanism | Experimental result | Reference |
|---|---|---|---|
| Physical | |||
| Physical barriers | Limit penetration of CDDP into tumors | Decreased CDDP accumulation in tumor cells | [62,63] |
| Fluidic shear stress | Activation of PI3K/Akt signaling and ABC drug transporters | Cancer stemness progression and CDDP resistance induced by fluidic shear stress | [64] |
| ECM | 1. Limited CDDP diffusion 2. The activation of survival signals through the interaction with tumor cells | Increased cancer cell sensitivity to CDDP in collagen- and fibronectin-deficient ECMs | [65] |
| Biological | |||
| Hypoxia | Increased cancer cell stemness and multidrug transporter expression | Increased CDDP resistance in low oxygen levels | [66,67,68,69,70] |
| Acidity | Increased multidrug transporter expression | Increased CDDP resistance in acidic conditions | [71,72] |
| CAF | 1. CAF-secreted growth factors or cytokines affecting cell apoptosis or intrinsic drug resistance 2. Metabolism of CAFs regulated by effector T-cells | 1. Increased CDDP resistance by CAF-secreted cytokines such as IL-6, IL-8, IL-11, insulin-like growth factor 1, and TGF-β 2. CAFs-mediated GSH metabolism and platinum resistance abrogated by cytotoxic T cells | [73,74,75,76,77,78] |
| TAM | Secretion of cytokines by TAM in an M2 polarization state | Increased CDDP resistance by TAM-secreted cytokines such as IL-6 and type I interferon | [79,80] |
| Drug | Category | Major target | Clinical benefit | Reference |
|---|---|---|---|---|
| Bevacizumab | Angiogenesis antagonist | Vascular endothelial growth factor A | 1. In the AVAil study, combination therapy (cisplatin, gemcitabine plus bevacizumab) prolonged PFS (HR = 0.82; p = 0.03) in first-line therapy for patients with advanced nonsquamous nonsmall-cell lung cancer compared with the control group (cisplatin plus gemcitabine). 2. In the AVAPERL study, combination therapy (cisplatin, pemetrexed plus bevacizumab) prolonged PFS (HR = 0.48; p < 0.001) in first-line therapy for patients with advanced nonsquamous nonsmall-cell lung cancer compared with the control group (cisplatin plus pemetrexed). | [84,85] |
| Pembrolizumab | Immune check point inhibitor | Programmed cell death protein 1 | In the KEYNOTE-189 study, combination therapy (cisplatin, pemetrexed plus pembrolizumab) increased OS at 12 months (HR = 0.49; p < 0.001) in first-line therapy for patients with advanced nonsmall-cell lung cancer compared with the control group (cisplatin plus pemetrexed). | [101] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. https://doi.org/10.3390/ijms20174136
Chen S-H, Chang J-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. International Journal of Molecular Sciences. 2019; 20(17):4136. https://doi.org/10.3390/ijms20174136
Chicago/Turabian StyleChen, Shang-Hung, and Jang-Yang Chang. 2019. "New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment" International Journal of Molecular Sciences 20, no. 17: 4136. https://doi.org/10.3390/ijms20174136
APA StyleChen, S. -H., & Chang, J. -Y. (2019). New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. International Journal of Molecular Sciences, 20(17), 4136. https://doi.org/10.3390/ijms20174136