Structural Analysis and Dynamic Processes of the Transmembrane Segment Inside Different Micellar Environments—Implications for the TM4 Fragment of the Bilitranslocase Protein

 ,
,  ,
, 


Abstract
:1. Introduction
2. Results
2.1. Prediction of the TM4 Transmembrane Region
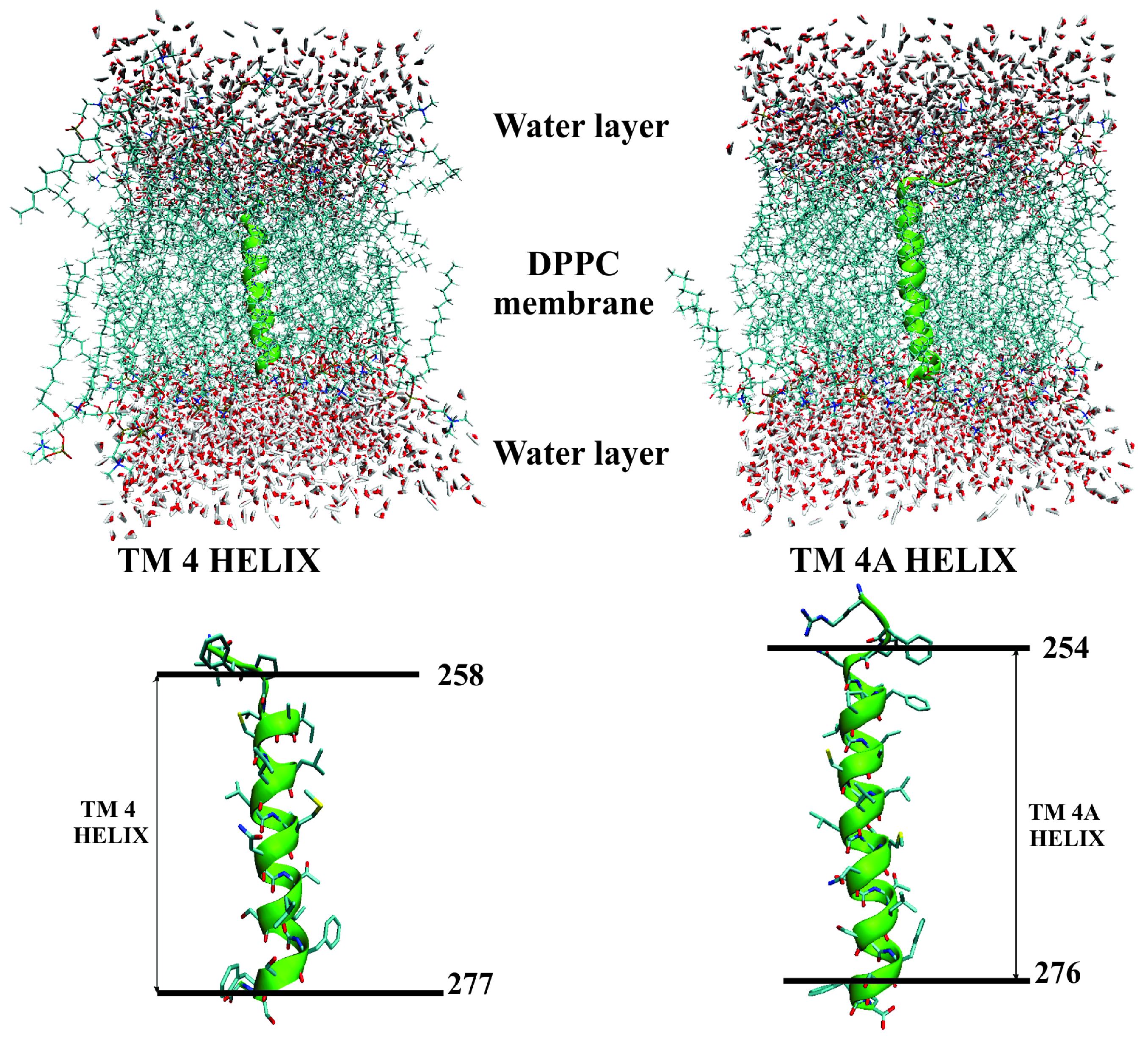
2.2. Assessment of the TM4/TM4A Stability in DPPC From MD Simulations
2.3. CD Spectroscopy Confirm Existence -Helical Conformation for the TM4 Fragment in SDS and DPC Surfactants
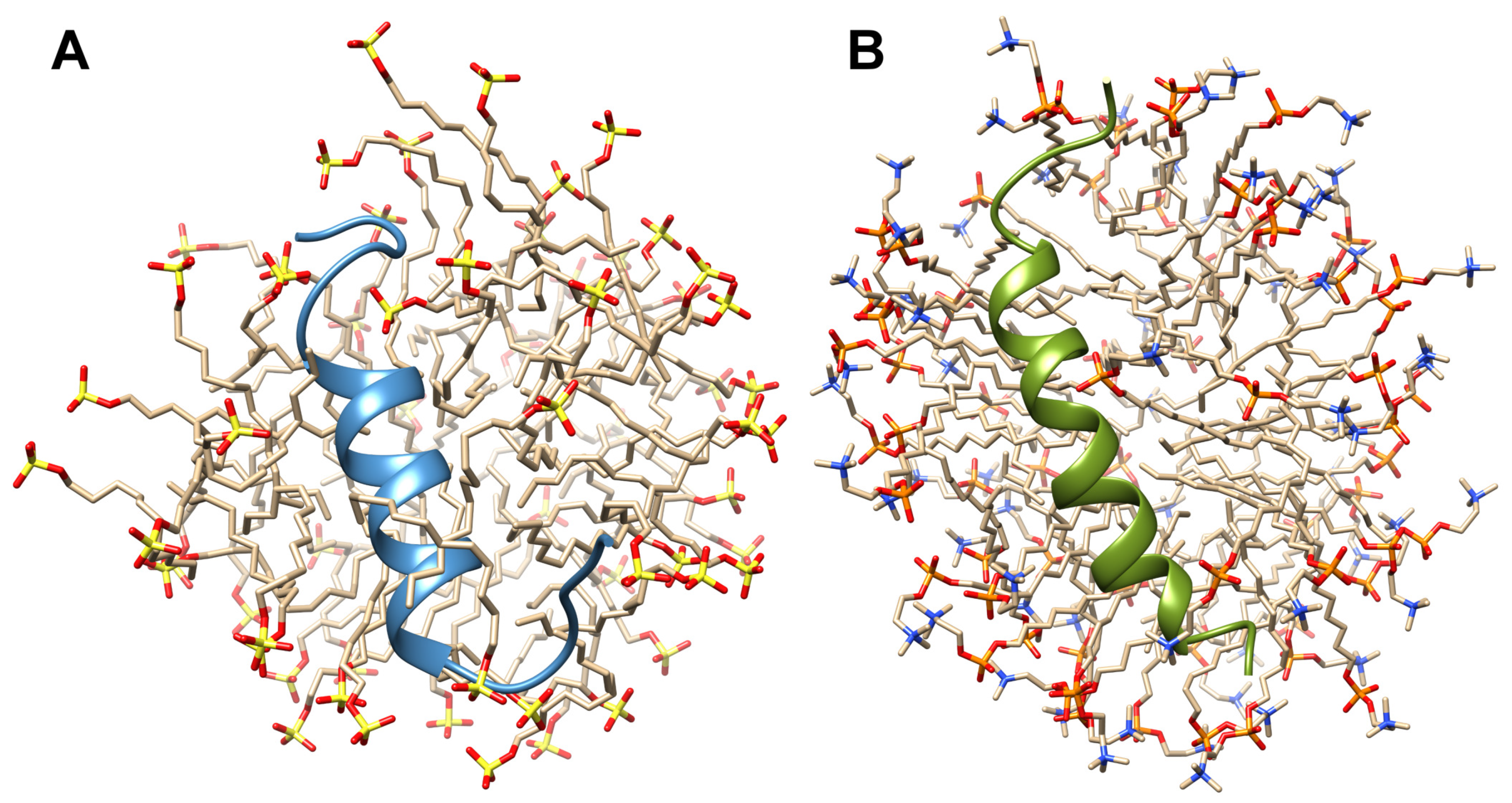
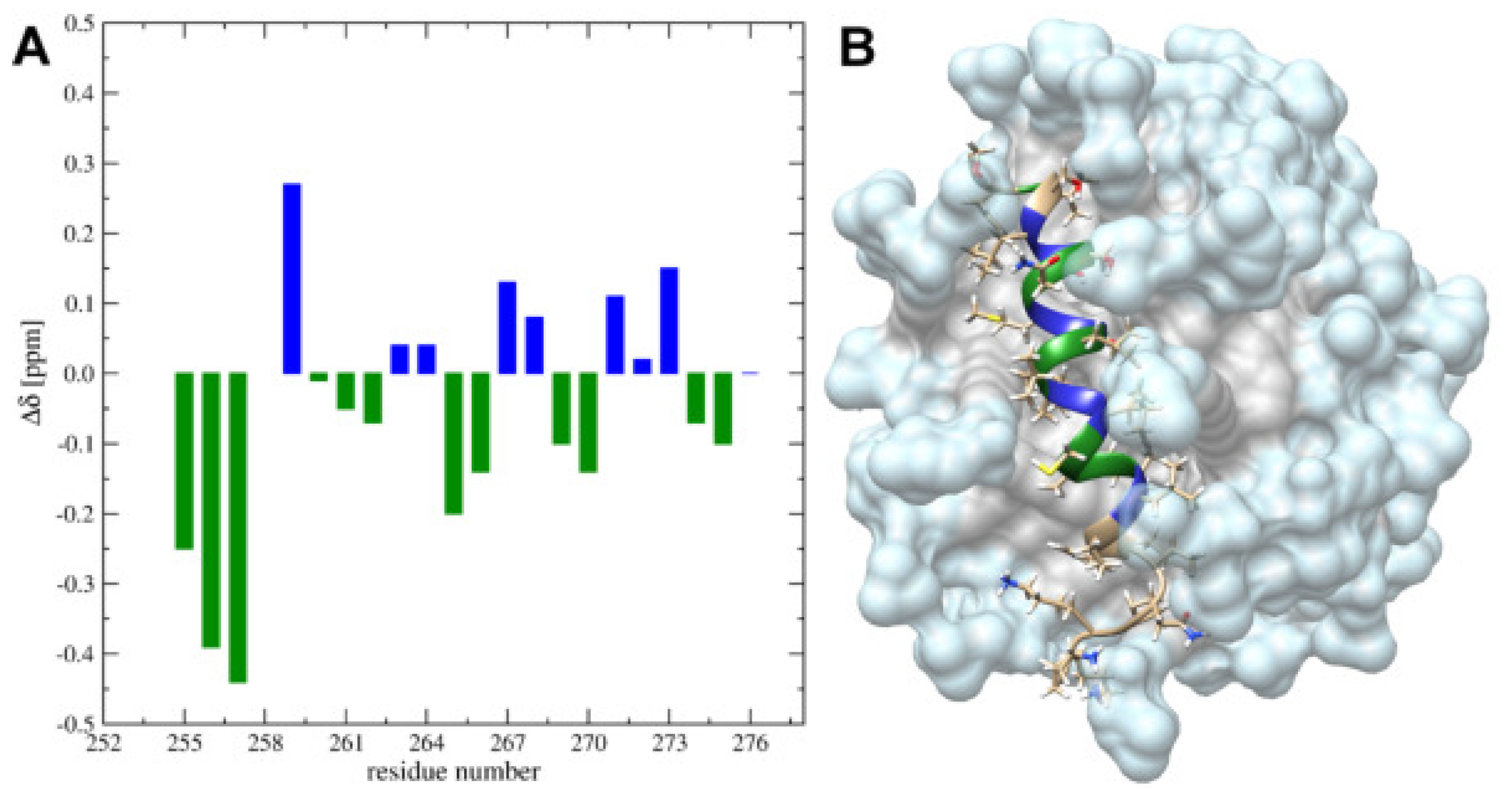
2.4. Solution High-Resolution 3D Structure of the TM4 Fragment in SDS- and DPC- Micelle by Means NMR Spectroscopy
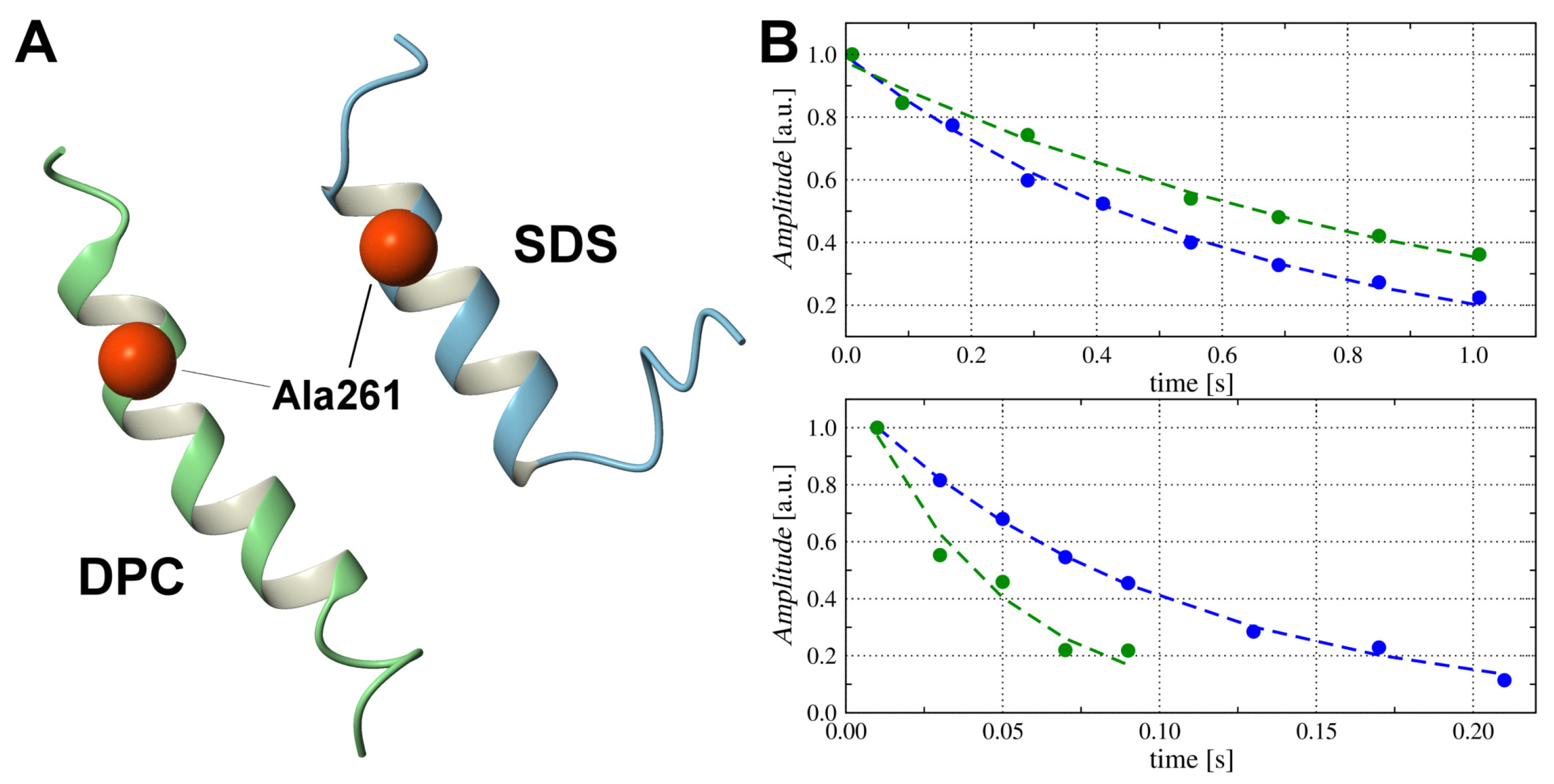
2.5. Translational Mobility of TM4 Fragments and Micellar Media with H, H and P PGSE NMR
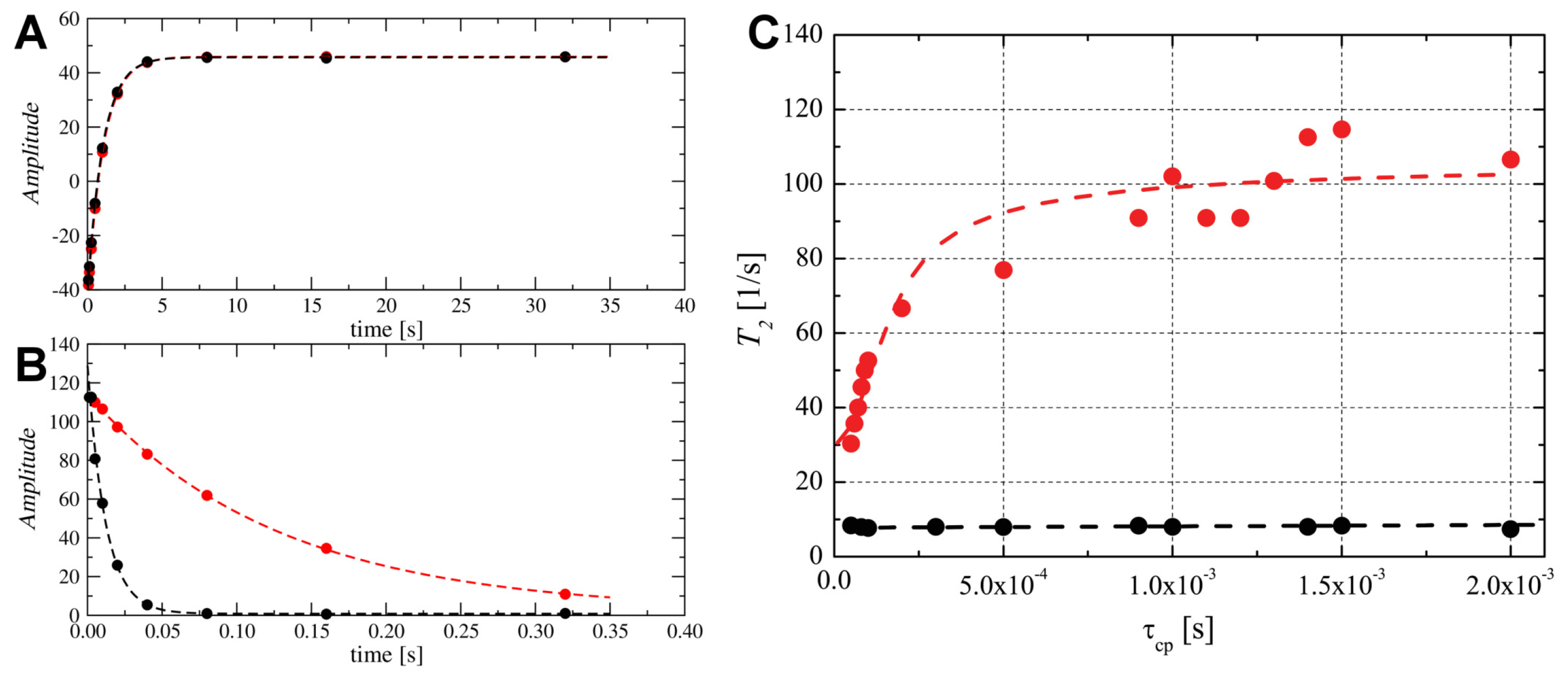
2.6. Probing the Molecular Dynamics of TM4 Segment Inside SDS- and DPC- Micelles by N NMR Relaxation
2.7. Snorkelling Interactions between N-Terminal Lysines and Phosphate Groups Observed in the DPC- Micelle by P NMR Relaxation Experiments
3. Discussion
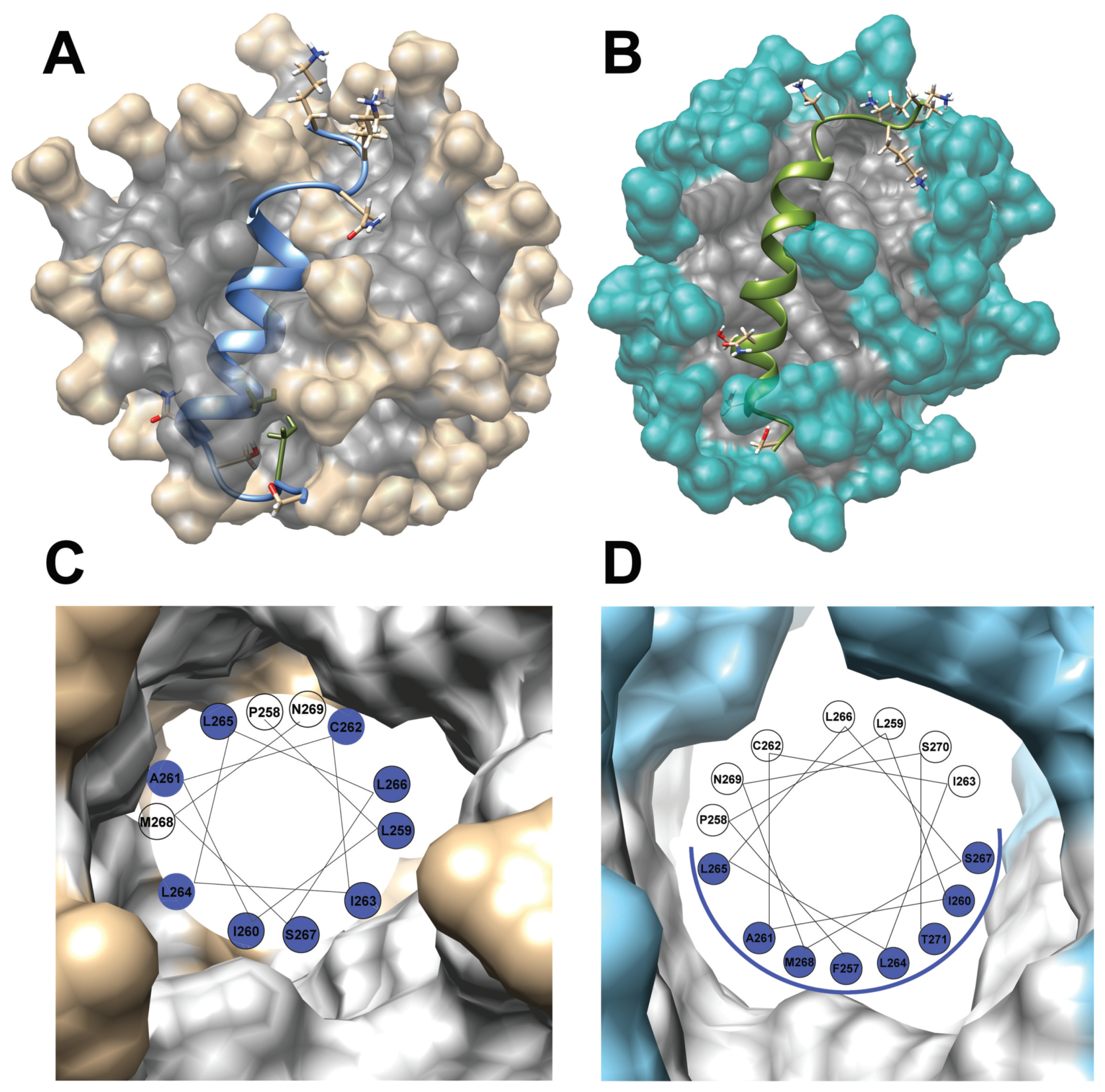
3.1. The Change from an Anionic to a Zwitterionic Environment Results in a Different Character of the -Helix and Backbone Dynamics
3.2. A Comparison with Previously Evaluated Structures of the TM2 and TM3 Transmembrane Fragments in SDS Micelle
4. Conclusions
5. Materials and Methods
5.1. Prediction Algorithm of the TM4 Transmembrane Segment
Molecular Dynamics (MD) Simulation Procedure of the TM4 Fragment in DPPC Lipid
5.2. Synthesis of the TM4 Peptide
5.3. 3D Structure of the TM4 Fragment in SDS and DPC Micelles with NMR Spectroscopy
5.4. H, H and P PGSE Diffusion Measurements of the TM4 Fragment in SDS- and DPC- Surfactants
5.5. Acquisition of N and P NMR Relaxation Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BTL | Bilitranslocase |
| TM | Trans-membrane |
| TM4 | Fourth TM4 region |
| CD | Circular dichoism |
| SDS | Sodium dodecyl sulfate |
| DPC | Dodecylphosphocholine |
| TOCSY | TOtal Correlation SpectroscopY |
| NOESY | Nuclear Overhauser Effect SpectroscopY |
| HSQC | Heteronuclear Single Quantum Correlation spectroscopy |
| DSS | Sodium 2,2-dimethyl-2-silapentane-5-sulfonate |
| CPMG | Carr-Purcell-Meiboom-Gill, PGSE—Pulsed Gradient Spin Echo |
| DPFGDSTE | Double Pulsed Field Gradient Double Stimulated Echo |
| RDF | Radial distribution function |
| CR | Caver-Richards model. |
References
- Tzakos, A.G.; Briasoulis, E.; Thalhammer, T.; Jäger, W.; Apostolopoulos, V. Novel oncology therapeutics: Targeted drug delivery for cancer. J. Drug Deliv. 2013, 2013, 918304. [Google Scholar] [CrossRef] [PubMed]
- Buxhofer-Ausch, V.; Secky, L.; Wlcek, K.; Svoboda, M.; Kounnis, V.; Briasoulis, E.; Tzakos, A.G.; Jaeger, W.; Thalhammer, T. Tumor-specific expression of organic anion-transporting polypeptides: transporters as novel targets for cancer therapy. J. Drug Deliv. 2013, 2013, 863539. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, S.; Terdoslavich, M.; Franca, R.; Vanzo, A.; Tramer, F.; Braidot, E.; Petrussa, E.; Vianello, A. Bioavailability of flavonoids: A review of their membrane transport and the function of bilitranslocase in animal and plant organisms. Curr. Drug Metab. 2009, 10, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Terdoslavich, M.; de Graaf, I.A.; Proost, J.H.; Cocolo, A.; Passamonti, S.; Groothuis, G.M. Bilitranslocase is involved in the uptake of bromosulfophthalein in rat and human liver. Drug Metab. Lett. 2012, 6, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, S.; Terdoslavich, M.; Margon, A.; Cocolo, A.; Medic, N.; Micali, F.; Decorti, G.; Franko, M. Uptake of bilirubin into HepG2 cells assayed by thermal lens spectroscopy. FEBS J. 2005, 272, 5522–5535. [Google Scholar] [CrossRef] [PubMed]
- Montanic, S.; Terdoslavich, M.; Rajcevic, U.; De Leo, L.; Curin Serbec, V.; Passamonti, S. Development and characterization of a novel mAb against bilitranslocase-a new biomarker of renal carcinoma. Radiol. Oncol. 2013, 47, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Župerl, Š.; Fornasaro, S.; Novič, M.; Passamonti, S. Experimental determination and prediction of bilitranslocase transport activity. Anal. Chim. Acta 2011, 705, 322–333. [Google Scholar] [CrossRef]
- Battiston, L.; Passamonti, S.; Macagno, A.; Sottocasa, G.L. The bilirubin-binding motif of bilitranslocase and its relation to conserved motifs in ancient biliproteins. Biochem. Biophys. Res. Commun. 1998, 247, 687–692. [Google Scholar] [CrossRef]
- Passamonti, S.; Battiston, L.; Sottocasa, G.L. Bilitranslocase can exist in two metastable forms with different affinities for the substrates. FEBS J. 1998, 253, 84–90. [Google Scholar]
- Karawajczyk, A.; Drgan, V.; Medic, N.; Oboh, G.; Passamonti, S.; Novič, M. Properties of flavonoids influencing the binding to bilitranslocase investigated by neural network modelling. Biochem. Pharmacol. 2007, 73, 308–320. [Google Scholar] [CrossRef]
- Passamonti, S.; Cocolo, A.; Braidot, E.; Petrussa, E.; Peresson, C.; Medic, N.; Macri, F.; Vianello, A. Characterization of electrogenic bromosulfophthalein transport in carnation petal microsomes and its inhibition by antibodies against bilitranslocase. FEBS J. 2005, 272, 3282–3296. [Google Scholar] [CrossRef] [Green Version]
- Maestro, A.; Terdoslavich, M.; Vanzo, A.; Kuku, A.; Tramer, F.; Nicolin, V.; Micali, F.; Decorti, G.; Passamonti, S. Expression of bilitranslocase in the vascular endothelium and its function as a flavonoid transporter. Cardiovasc. Res. 2009, 85, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Ziberna, L.; Tramer, F.; Moze, S.; Vrhovsek, U.; Mattivi, F.; Passamonti, S. Transport and bioactivity of cyanidin 3-glucoside into the vascular endothelium. Free Radic. Biol. Med. 2012, 52, 1750–1759. [Google Scholar] [CrossRef]
- Roy Choudhury, A.; Novič, M. Data-driven model for the prediction of protein transmembrane regions. SAR QSAR Environ. Res. 2009, 20, 741–754. [Google Scholar] [CrossRef]
- Perdih, A.; Choudhury, A.R.; Župerl, Š.; Sikorska, E.; Zhukov, I.; Solmajer, T.; Novič, M. Structural analysis of a peptide fragment of transmembrane transporter protein bilitranslocase. PLoS ONE 2012, 7, e38967. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Perdih, A.; Župerl, Š.; Sikorska, E.; Solmajer, T.; Jurga, S.; Zhukov, I.; Novič, M. Structural elucidation of transmembrane transporter protein Bilitranslocase: Conformational analysis of the second transmembrane region TM2 by molecular dynamics and NMR spectroscopy. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2609–2619. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Sikorska, E.; van den Boom, J.; Bayer, P.; Popenda, Ł.; Szutkowski, K.; Jurga, S.; Bonomi, M.; Sali, A.; Zhukov, I.; et al. Structural model of the bilitranslocase transmembrane domain supported by NMR and FRET data. PLoS ONE 2015, 10, e0135455. [Google Scholar] [CrossRef]
- Pieper, U.; Schlessinger, A.; Kloppmann, E.; Chang, G.A.; Chou, J.J.; Dumont, M.E.; Fox, B.G.; Fromme, P.; Hendrickson, W.A.; Malkowski, M.G.; et al. Coordinating the impact of structural genomics on the human α-helical transmembrane proteome. Nat. Struct. Mol. Biol. 2013, 20, 135–138. [Google Scholar] [CrossRef]
- Vashisth, H.; Storaska, A.J.; Neubig, R.R.; Brooks, C.L., III. Conformational dynamics of a regulator of G-protein signaling protein reveals a mechanism of allosteric inhibition by a small molecule. ACS Chem. Biol. 2013, 8, 2778–2784. [Google Scholar] [CrossRef]
- Schmidpeter, P.A.; Schmid, F.X. Molecular determinants of a regulatory prolyl isomerization in the signal adapter protein c-CrkII. ACS Chem. Biol. 2014, 9, 1145–1152. [Google Scholar] [CrossRef]
- Nugent, T.; Jones, D.T. Membrane protein structural bioinformatics. J. Struct. Biol. 2012, 179, 327–337. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Novič, M. PredβTM: A novel β-transmembrane region prediction algorithm. PLoS ONE 2015, 10, e0145564. [Google Scholar] [CrossRef]
- Nieh, M.P.; Raghunathan, V.A.; Kline, S.R.; Harroun, T.A.; Huang, C.Y.; Pencer, J.; Katsaras, J. Spontaneously formed unilamellar vesicles with path-dependent size distribution. Langmuir 2005, 21, 6656–6661. [Google Scholar] [CrossRef]
- Kallick, D.A.; Tessmer, M.R.; Watts, C.R.; Li, C.Y. The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. Ser. B 1995, 109, 60–65. [Google Scholar] [CrossRef]
- Chipot, C.; Dehez, F.; Schnell, J.R.; Zitzmann, N.; Pebay-Peyroula, E.; Catoire, L.J.; Miroux, B.; Kunji, E.R.; Veglia, G.; Cross, T.A.; et al. Perturbations of native membrane protein structure in alkyl phosphocholine detergents: A critical assessment of NMR and biophysical studies. Chem. Rev. 2018, 118, 3559–3607. [Google Scholar] [CrossRef]
- Zhou, H.X.; Cross, T.A. Modeling the membrane environment has implications for membrane protein structure and function: Influenza A M2 protein. Protein Sci. 2013, 22, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, K. TMbase-A database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 1993, 374, 166. [Google Scholar]
- Claros, M.G.; von Heijne, G. TopPred II: An improved software for membrane protein structure predictions. Bioinformatics 1994, 10, 685–686. [Google Scholar] [CrossRef]
- Pasquier, C.; Promponas, V.; Palaios, G.; Hamodrakas, J.; Hamodrakas, S. A novel method for predicting transmembrane segments in proteins based on a statistical analysis of the SwissProt database: The PRED-TMR algorithm. Protein Eng. 1999, 12, 381–385. [Google Scholar] [CrossRef]
- Shen, H.; Chou, J.J. MemBrain: Improving the accuracy of predicting transmembrane helices. PLoS ONE 2008, 3, e2399. [Google Scholar] [CrossRef]
- Reynolds, S.M.; Käll, L.; Riffle, M.E.; Bilmes, J.A.; Noble, W.S. Transmembrane topology and signal peptide prediction using dynamic bayesian networks. PLoS Comput. Biol. 2008, 4, e1000213. [Google Scholar] [CrossRef]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Bernsel, A.; Viklund, H.; Falk, J.; Lindahl, E.; von Heijne, G.; Elofsson, A. Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. USA 2008, 105, 7177–7181. [Google Scholar] [CrossRef] [Green Version]
- Bernsel, A.; Viklund, H.; Hennerdal, A.; Elofsson, A. TOPCONS: Consensus prediction of membrane protein topology. Nucleic Acids Res. 2009, 37, W465–W468. [Google Scholar] [CrossRef]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Page, R.C.; Kim, S.; Cross, T.A. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure 2008, 16, 787–797. [Google Scholar] [CrossRef]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef]
- Baldwin, R.L. Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. USA 1986, 83, 8069–8072. [Google Scholar] [CrossRef]
- Mu, Y.; Gao, Y.Q. Effects of hydrophobic and dipole-dipole interactions on the conformational transitions of a model polypeptide. J. Chem. Phys. 2007, 127, 09B604. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 2004, 13, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Miles, A.J.; Wallace, B.A. Circular dichroism spectroscopy of membrane proteins. Chem. Soc. Rev. 2016, 45, 4859–4872. [Google Scholar] [CrossRef] [Green Version]
- Wüthrich, K. NMR with Proteins and Nucleic Acids. Europhys. News 1986, 17, 11–13. [Google Scholar] [CrossRef]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. In Artificial Neural Networks; Springer: New York, NY, USA, 2015; pp. 17–32. [Google Scholar]
- Kholodenko, A.L.; Douglas, J.F. Generalized Stokes-Einstein equation for spherical particle suspensions. Phys. Rev. E 1995, 51, 1081. [Google Scholar] [CrossRef]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry, Part 2: Techniques for the Study of Biological Structure and Function, Part 2; Freeman: San Francisco, CA, USA, 1980. [Google Scholar]
- Chou, J.J.; Baber, J.L.; Bax, A. Characterization of phospholipid mixed micelles by translational diffusion. J. Biomol. NMR 2004, 29, 299–308. [Google Scholar] [CrossRef]
- Beswick, V.; Guerois, R.; Cordier-Ochsenbein, F.; Coic, Y.M.; Huynh-Dinh, T.; Tostain, J.; Noel, J.P.; Sanson, A.; Neumann, J.M. Dodecylphosphocholine micelles as a membrane-like environment: New results from NMR relaxation and paramagnetic relaxation enhancement analysis. Eur. Biophys. J. 1998, 28, 48–58. [Google Scholar] [CrossRef]
- Jafari, M.; Mehrnejad, F.; Doustdar, F. Insight into the interactions, residue snorkeling, and membrane disordering potency of a single antimicrobial peptide into different lipid bilayers. PLoS ONE 2017, 12, e0187216. [Google Scholar] [CrossRef]
- Szutkowski, K.; Stilbs, P.; Jurga, S. Proton chemical exchange in aqueous solutions of dodecylammonium chloride: Effects of micellar aggregation. J. Phys. Chem. C 2007, 111, 15613–15619. [Google Scholar] [CrossRef]
- Carver, J.; Richards, R. A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J. Magn. Reson. 1972, 6, 89–105. [Google Scholar] [CrossRef]
- Palmer, A.G., III; Kroenke, C.D.; Loria, J.P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001, 339, 204–238. [Google Scholar]
- Langham, A.A.; Waring, A.J.; Kaznessis, Y. Comparison of interactions between beta-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochem. 2007, 8, 11. [Google Scholar] [CrossRef]
- Venko, K.; Choudhury, A.R.; Novič, M. Computational approaches for revealing the structure of membrane transporters: Case study on bilitranslocase. Comput. Struct. Biotechnol. J. 2017, 15, 232–242. [Google Scholar] [CrossRef]
- Nogales, D.; Lightner, D.A. On the structure of bilirubin in solution. J. Biol. Chem. 1995, 270, 73–77. [Google Scholar] [CrossRef]
- Xie, M.; Holmes, D.L.; Lightner, D.A. Bilirubin conformation and intramolecular steric buttressing. C(10)-gem-dimethyl effect. Tetrahedron 1993, 49, 9235–9250. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Yuan, Z.; Mattick, J.S.; Teasdale, R.D. SVMtm: Support vector machines to predict transmembrane segments. J. Comput. Chem. 2004, 25, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Cserzo, M.; Eisenhaber, F.; Eisenhaber, B.; Simon, I. TM or not TM: Transmembrane protein prediction with low false positive rate using DAS-TMfilter. Bioinformatics 2004, 20, 136–137. [Google Scholar] [CrossRef]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar] [CrossRef]
- Viklund, H.; Elofsson, A. A method that improves topology prediction for transmembrane proteins by using two-track ANN-based preference scores and an improved topological grammar. Bioinformatics 2008, 24, 1662–1668. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- MacKerell, A., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R., Jr.; Evanseck, J.; Field, M.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Racine, J. gnuplot 4.0: A portable interactive plotting utility. J. Appl. Econom. 2006, 21, 133–141. [Google Scholar] [CrossRef]
- Turro, N.J.; Yekta, A. Luminescent probes for detergent solutions. A simple procedure for determination of the mean aggregation number of micelles. J. Am. Chem. Soc. 1978, 100, 5951–5952. [Google Scholar] [CrossRef]
- Hsu, S.T.D.; Breukink, E.; Bierbaum, G.; Sahl, H.G.; de Kruijff, B.; Kaptein, R.; van Nuland, N.A.; Bonvin, A.M. NMR study of Mersacidin and lipid II interaction in dodecylphosphocholine micelles. J. Biol. Chem. 2003, 278, 13110–13117. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2014, 31, 1325–1327. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 2010, 46, 199–204. [Google Scholar] [CrossRef]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.; Cheatham, T.E., III; Simmerling, C.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Pearlman, D.A.; Crowley, M.; et al. AMBER 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Nilsson, M.; Gil, A.M.; Delgadillo, I.; Morris, G.A. Improving pulse sequences for 3D diffusion-ordered NMR spectroscopy: 2DJ-IDOSY. Anal. Chem. 2004, 76, 5418–5422. [Google Scholar] [CrossRef]
- Wu, D.; Chen, A.; Johnson, C.S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Scotti, A.; Liu, W.; Hyatt, J.; Herman, E.; Choi, H.; Kim, J.; Lyon, L.; Gasser, U.; Fernandez-Nieves, A. The CONTIN algorithm and its application to determine the size distribution of microgel suspensions. J. Chem. Phys. 2015, 142, 234905. [Google Scholar] [CrossRef] [Green Version]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Kay, L.E.; Nicholson, L.K.; Delaglio, F.; Bax, A.; Torchia, D.A. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 1992, 97, 359–375. [Google Scholar] [CrossRef]
- Li, C.; Gao, P.; Qin, H.; Chase, R.; Gor’kov, P.L.; Brey, W.W.; Cross, T.A. Uniformly aligned full-length membrane proteins in liquid crystalline bilayers for structural characterization. J. Am. Chem. Soc. 2007, 129, 5304–5305. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Media | R (s) | R (s) | (ns) |
|---|---|---|---|
| SDS-d | 1.59 ± 0.08 | 10.01 ± 0.81 | 7.26 ± 0.41 |
| DPC-d | 0.68 ± 0.01 | 28.58 ± 1.09 | 20.56 ± 0.41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szutkowski, K.; Sikorska, E.; Bakanovych, I.; Choudhury, A.R.; Perdih, A.; Jurga, S.; Novič, M.; Zhukov, I. Structural Analysis and Dynamic Processes of the Transmembrane Segment Inside Different Micellar Environments—Implications for the TM4 Fragment of the Bilitranslocase Protein. Int. J. Mol. Sci. 2019, 20, 4172. https://doi.org/10.3390/ijms20174172
Szutkowski K, Sikorska E, Bakanovych I, Choudhury AR, Perdih A, Jurga S, Novič M, Zhukov I. Structural Analysis and Dynamic Processes of the Transmembrane Segment Inside Different Micellar Environments—Implications for the TM4 Fragment of the Bilitranslocase Protein. International Journal of Molecular Sciences. 2019; 20(17):4172. https://doi.org/10.3390/ijms20174172
Chicago/Turabian StyleSzutkowski, Kosma, Emilia Sikorska, Iulia Bakanovych, Amrita Roy Choudhury, Andrej Perdih, Stefan Jurga, Marjana Novič, and Igor Zhukov. 2019. "Structural Analysis and Dynamic Processes of the Transmembrane Segment Inside Different Micellar Environments—Implications for the TM4 Fragment of the Bilitranslocase Protein" International Journal of Molecular Sciences 20, no. 17: 4172. https://doi.org/10.3390/ijms20174172
APA StyleSzutkowski, K., Sikorska, E., Bakanovych, I., Choudhury, A. R., Perdih, A., Jurga, S., Novič, M., & Zhukov, I. (2019). Structural Analysis and Dynamic Processes of the Transmembrane Segment Inside Different Micellar Environments—Implications for the TM4 Fragment of the Bilitranslocase Protein. International Journal of Molecular Sciences, 20(17), 4172. https://doi.org/10.3390/ijms20174172