Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies

Abstract
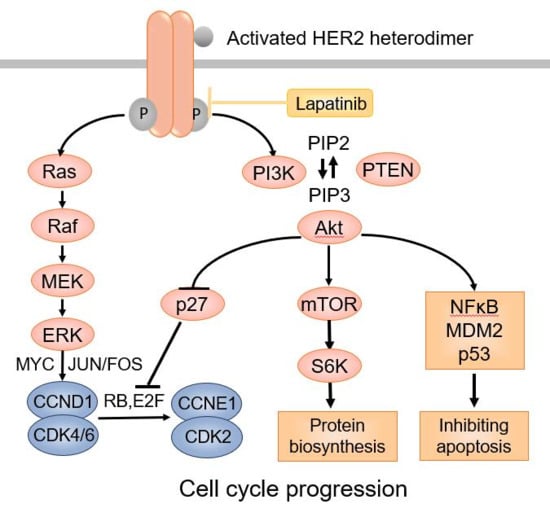
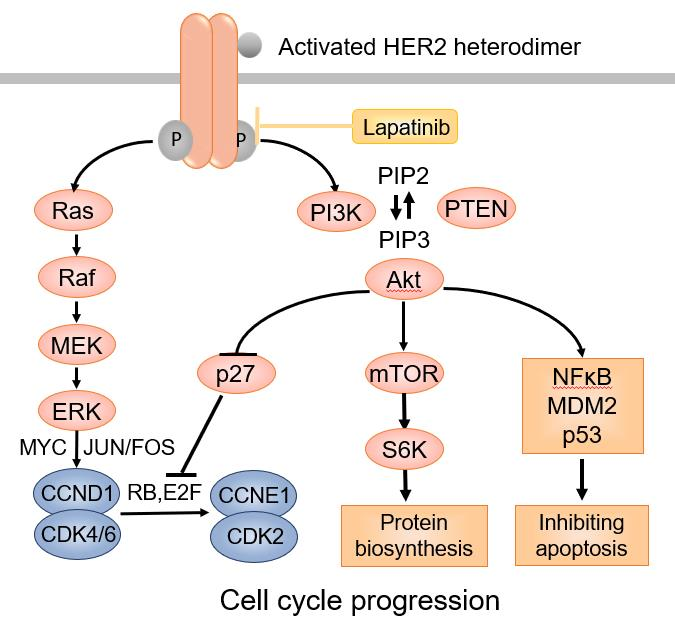
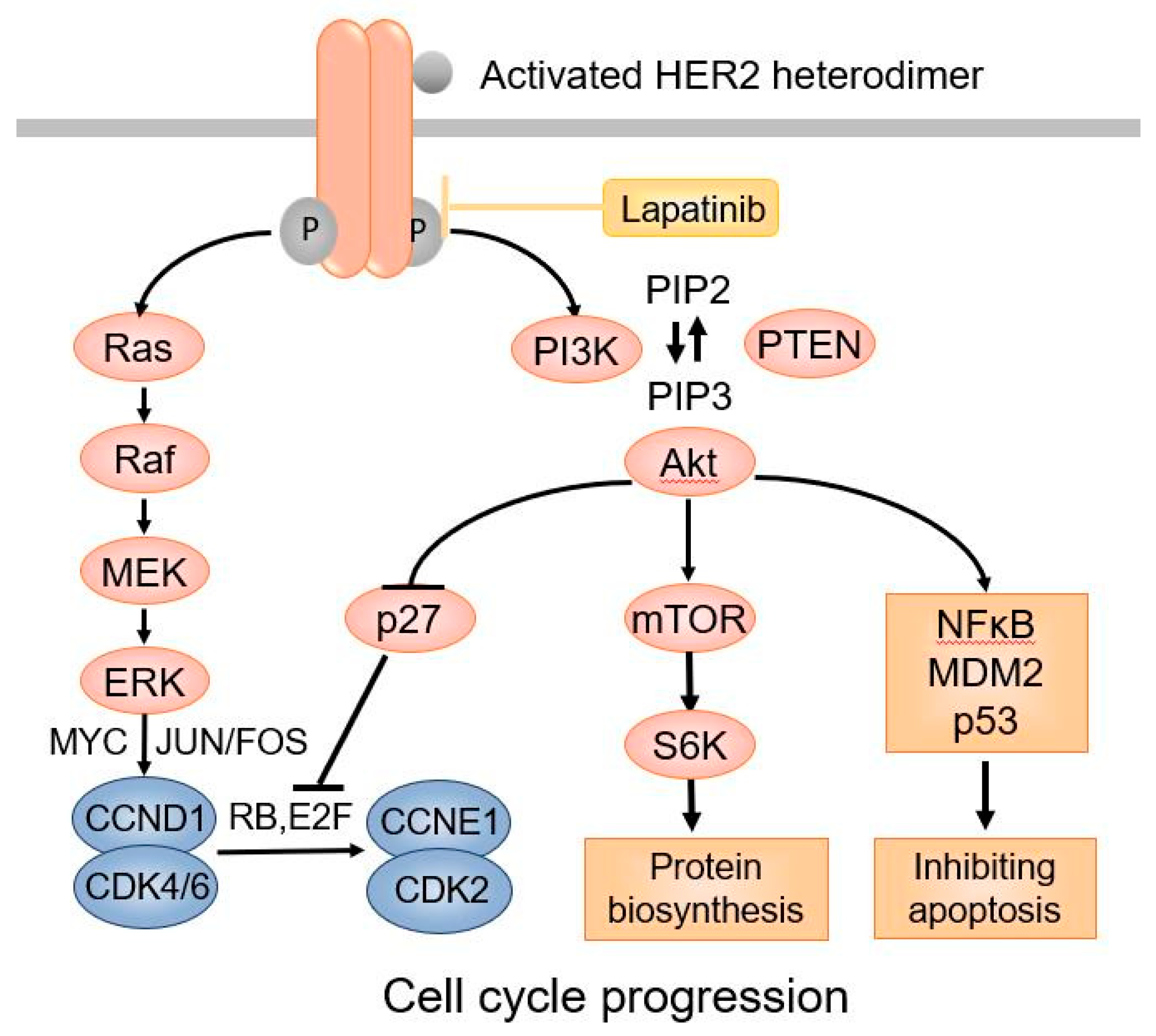
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Human Epidermal Growth Factor Receptor 2 (HER2) Expression in OS
3. Association of Trastuzumab (TRA) with Graphene Oxide (GO]
3.1. TRA Can Stably Associate with Graphene Oxide (GO) through Noncovalent Bonds
3.2. GO-Associated TRA Demonstrated Enhances Binding to HER2lo OS Cells
3.3. TRA/GO Is Cytotoxic to OS Cells
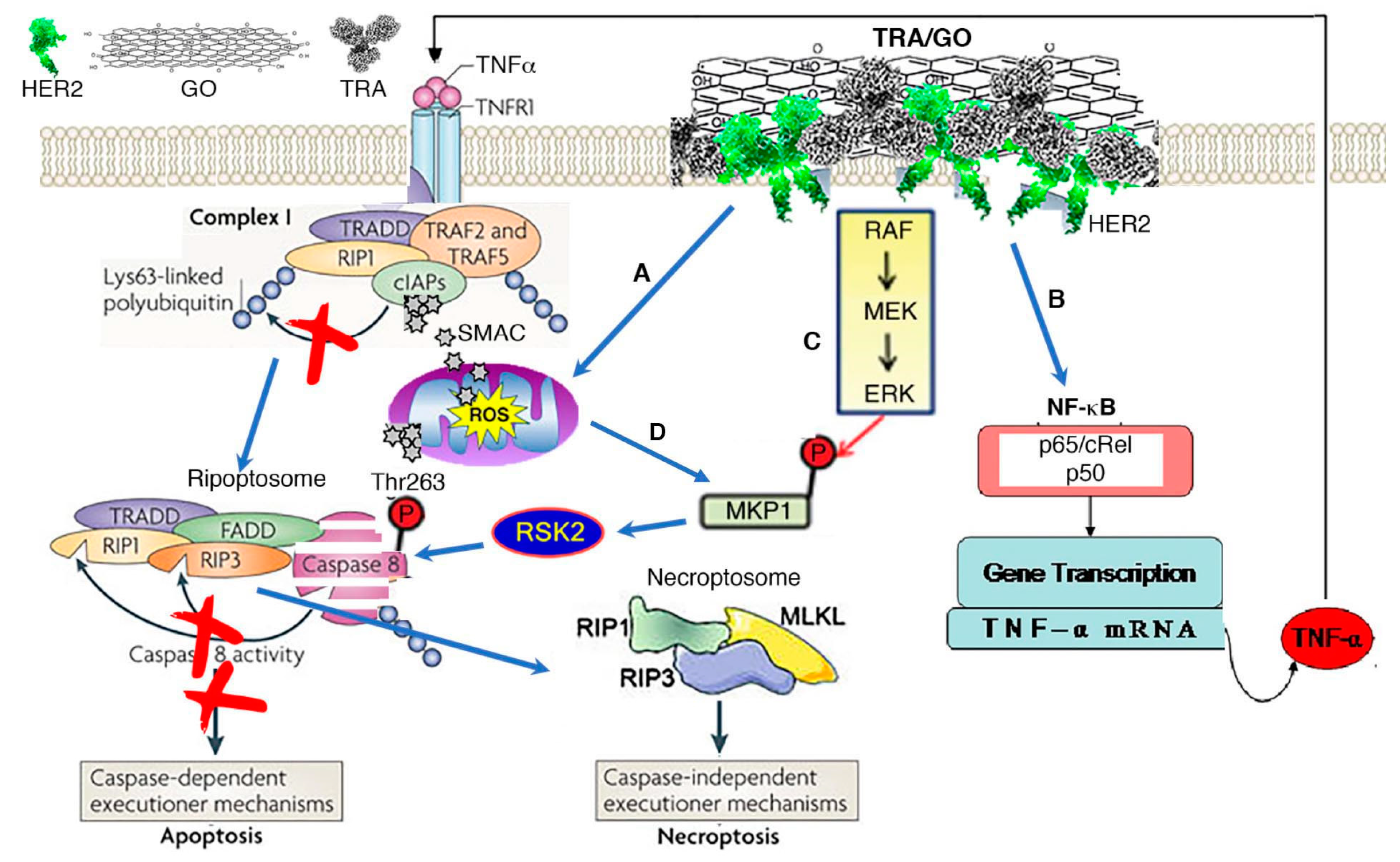
3.4. TRA/GO Induces Oxidative Stress as well as HER2 Signaling in OS Cells
3.5. TRA/GO Kills OS by Necroptosis
3.6. TRA/GO Eliminate Established Xenograft OS
4. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT J. 2018, 4, 12. [Google Scholar] [CrossRef]
- Ozaki, T.; Flege, S.; Liljenqvist, U.; Hillmann, A.; Delling, G.; Salzer-Kuntschik, M.; Jürgens, H.; Kotz, R.; Winkelmann, W.; Bielack, S.S. Osteosarcoma of the spine: Experience of the Cooperative Osteosarcoma Study Group. Cancer 2002, 94, 1069–1077. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Cadoo, K.A.; Walsh, E.M.; Emerson, R.; Dervan, P.; O’Keane, C.; Hurson, B.; O’Toole, G.; Dudeney, S.; Kavanagh, E.; et al. Perioperative chemotherapy in the treatment of osteosarcoma: A 26-year single institution review. Clin. Sarcoma Res. 2015, 5, 17. [Google Scholar] [CrossRef]
- Bacci, G.; Ferrari, S.; Longhi, A.; Forni, C.; Bertoni, F.; Fabbri, N.; Zavatta, M.; Versari, M. Neoadjuvant chemotherapy for high grade osteosarcoma of the extremities: Long-term results for patients treated according to the Rizzoli IOR/OS-3b protocol. J. Chemother. 2001, 13, 93–99. [Google Scholar] [CrossRef]
- Schwartz, C.L.; Wexler, L.H.; Krailo, M.D.; Teot, L.A.; Devidas, M.; Steinherz, L.J.; Goorin, A.M.; Gebhardt, M.C.; Healey, J.H.; Sato, J.K.; et al. Intensified Chemotherapy With Dexrazoxane Cardioprotection in Newly Diagnosed Nonmetastatic Osteosarcoma: A Report From the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 54–61. [Google Scholar] [CrossRef]
- He, H.; Ni, J.; Huang, J. Molecular mechanisms of chemoresistance in osteosarcoma (Review). Oncol. Lett. 2014, 7, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Messerschmitt, P.J.; Rettew, A.N.; Brookover, R.E.; Garcia, R.M.; Getty, P.J.; Greenfield, E.M. Specific tyrosine kinase inhibitors regulate human osteosarcoma cells in vitro. Clin. Orthop. Relat. Res. 2008, 466, 2168–2175. [Google Scholar] [CrossRef]
- Pommier, Y.; Sordet, O.; Antony, S.; Hayward, R.L.; Kohn, K.W. Apoptosis defects and chemotherapy resistance: Molecular interaction maps and networks. Oncogene 2004, 23, 2934–2949. [Google Scholar] [CrossRef]
- Chen, R.; Wang, G.; Zheng, Y.; Hua, Y.; Cai, Z. Drug resistance-related microRNAs in osteosarcoma: Translating basic evidence into therapeutic strategies. J. Cell Mol. Med. 2019, 23, 2280–2292. [Google Scholar] [CrossRef]
- Sekar, D.; Mani, P.; Biruntha, M.; Sivagurunathan, P.; Karthigeyan, M. Dissecting the functional role of microRNA 21 in osteosarcoma. Cancer Gene. Ther. 2019, 26, 179–182. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Li, B.; Wang, S.; Chen, T.; Ye, Z. Innate Immune Cells: A Potential and Promising Cell Population for Treating Osteosarcoma. Front. Immunol. 2019, 10, 1114. [Google Scholar] [CrossRef]
- Roberts, S.S.; Chou, A.J.; Cheung, N.K.V. Immunotherapy of Childhood Sarcomas. Front. Oncol. 2015, 5, 181. [Google Scholar] [CrossRef] [Green Version]
- Wedekind, M.F.; Wagner, L.M.; Cripe, T.P. Immunotherapy for osteosarcoma: Where do we go from here? Pediatr. Blood Cancer 2018, 65, e27227. [Google Scholar] [CrossRef] [Green Version]
- Jimmy, R.; Stern, C.; Lisy, K.; White, S. Effectiveness of mifamurtide in addition to standard chemotherapy for high-grade osteosarcoma: A systematic review. JBI Database System. Rev. Implement. Rep. 2017, 15, 2113–2152. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, S3–S8. [Google Scholar] [CrossRef]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Brennan, P.J.; Kumogai, T.; Berezov, A.; Murali, R.; Greene, M.I. HER2/neu: Mechanisms of dimerization/oligomerization. Oncogene 2000, 19, 6093–6101. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Dori, S.; Vered, M.; David, R.; Buchner, A. HER2/neu expression in adenoid cystic carcinoma of salivary gland origin: An immunohistochemical study. J. Oral Pathol. Med. 2002, 31, 463–467. [Google Scholar] [CrossRef]
- Latif, Z.; Watters, A.D.; Bartlett, J.M.S.; Underwood, M.A.; Aitchison, M. Gene amplification and overexpression of HER2 in renal cell carcinoma. BJU Int. 2002, 89, 5–9. [Google Scholar] [CrossRef]
- Latif, Z.; Watters, A.D.; Dunn, I.; Grigor, K.; Underwood, M.A.; Bartlett, J.M.S. HER2/neu gene amplification and protein overexpression in G3 pT2 transitional cell carcinoma of the bladder: A role for anti-HER2 therapy? Eur. J. Cancer 2004, 40, 56–63. [Google Scholar] [CrossRef]
- Morris, M.J.; Reuter, V.E.; Kelly, W.K.; Slovin, S.F.; Kenneson, K.; Verbel, D.; Osman, I.; Scher, H.I. HER-2 profiling and targeting in prostate carcinoma. Cancer 2002, 94, 980–986. [Google Scholar] [CrossRef]
- Safran, H.; Steinhoff, M.; Mangray, S.; Rathore, R.; King, T.C.; Chai, L.; Berzein, K.; Moore, T.; Iannitti, D.; Reiss, P.; et al. Overexpression of the HER-2/neu oncogene in pancreatic adenocarcinoma. Am. J. Clin. Oncol. 2001, 24, 496–499. [Google Scholar] [CrossRef]
- Mar, N.; Vredenburgh, J.J.; Wasser, J.S. Targeting HER2 in the treatment of non-small cell lung cancer. Lung Cancer 2015, 87, 220–225. [Google Scholar] [CrossRef]
- Chavez-Blanco, A.; Perez-Sanchez, V.; Gonzalez-Fierro, A.; Vela-Chavez, T.; Candelaria, M.; Cetina, L.; Vidal, S.; Dueñas-Gonzalez, A. HER2 expression in cervical cancer as a potential therapeutic target. BMC Cancer 2004, 4, 59. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Morrison, C.; Zanagnolo, V.; Ramirez, N.; Cohn, D.E.; Kelbick, N.; Copeland, L.; Maxwell, L.G.; Fowler, J.M. HER-2 is an independent prognostic factor in endometrial cancer: Association with outcome in a large cohort of surgically staged patients. J. Clin. Oncol. 2006, 24, 2376–2385. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kim, S.; Choi, Y.L.; Cho, Y.J.; Oh, E.; Choi, J.J.; Jung, K.; Song, J.Y.; Ahn, S.E.; Kim, B.G.; et al. HER2 as a novel therapeutic target for cervical cancer. Oncotarget 2015, 6, 36219–36230. [Google Scholar] [CrossRef]
- Pollock, N.I.; Grandis, J.R. HER2 as a therapeutic target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 526–533. [Google Scholar] [CrossRef]
- Seigel, G.M.; Sharma, S.; Hackam, A.S.; Shah, D.K. HER2/ERBB2 immunoreactivity in human retinoblastoma. Tumour Biol. 2016, 37, 6135–6142. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch. Pathol. Lab. Med. 2014, 138, 241–256. [Google Scholar] [CrossRef]
- Baselga, J. Herceptin alone or in combination with chemotherapy in the treatment of HER2-positive metastatic breast cancer: Pivotal trials. Oncology 2001, 61 (Suppl. 2), 14–21. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef]
- Gorlick, R.; Huvos, A.G.; Heller, G.; Aledo, A.; Beardsley, G.P.; Healey, J.H.; Meyers, P.A. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J. Clin. Oncol. 1999, 17, 2781–2788. [Google Scholar] [CrossRef]
- Onda, M.; Matsuda, S.; Higaki, S.; Iijima, T.; Fukushima, J.I.; Yokokura, A.; Kojima, T.; Horiuchi, H.; Kurokawa, T.; Yamamoto, T. ErbB-2 expression is correlated with poor prognosis for patients with osteosarcoma. Cancer 1996, 77, 71–78. [Google Scholar] [CrossRef]
- Fellenberg, J.; Krauthoff, A.; Pollandt, K.; Delling, G.; Parsch, D. Evaluation of the predictive value of Her-2/neu gene expression on osteosarcoma therapy in laser-microdissected paraffin-embedded tissue. Lab. Investig. 2004, 84, 113–121. [Google Scholar] [CrossRef]
- Thomas, D.G.; Giordano, T.J.; Sanders, D.; Biermann, J.S.; Baker, L. Absence of HER2/neu gene expression in osteosarcoma and skeletal Ewing’s sarcoma. Clin. Cancer Res. 2002, 8, 788–793. [Google Scholar]
- Willmore-Payne, C.; Holden, J.A.; Zhou, H.; Gupta, D.; Hirschowitz, S.; Wittwer, C.T.; Layfield, L.J. Evaluation of Her-2/neu gene status in osteosarcoma by fluorescence in situ hybridization and multiplex and monoplex polymerase chain reactions. Arch. Pathol. Lab. Med. 2006, 130, 691–698. [Google Scholar]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef]
- Mason, N.J.; Gnanandarajah, J.S.; Engiles, J.B.; Gray, F.; Laughlin, D.; Gaurnier-Hausser, A.; Wallecha, A.; Huebner, M.; Paterson, Y. Immunotherapy with a HER2-Targeting Listeria Induces HER2-Specific Immunity and Demonstrates Potential Therapeutic Effects in a Phase I Trial in Canine Osteosarcoma. Clin. Cancer Res. 2016, 22, 4380–4390. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Peters, S.; Stahel, R.; Bubendorf, L.; Bonomi, P.; Villegas, A.; Kowalski, D.M.; Baik, C.S.; Isla, D.; Carpeno, J.D.C.; Garrido, P.; et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin. Cancer Res. 2019, 25, 64–72. [Google Scholar] [CrossRef]
- Büchler, P.; Reber, H.A.; Bückler, M.C.; Roth, M.A.; Büchler, M.W.; Friess, H.; Isacoff, W.H.; Hines, O.J. Therapy for pancreatic cancer with a recombinant humanized anti-HER2 antibody (herceptin). J. Gastrointest. Surg. 2001, 5, 139–146. [Google Scholar] [CrossRef]
- Mihaljevic, A.; Büchler, P.; Harder, J.; Hofheinz, R.; Gregor, M.; Kanzler, S.; Schmiegel, W.; Heinemann, V.; Endlicher, E.; Klöppel, G.; et al. A prospective, non-randomized phase II trial of Trastuzumab and Capecitabine in patients with HER2 expressing metastasized pancreatic cancer. BMC Surg. 2009, 9, 1. [Google Scholar] [CrossRef]
- Harder, J.; Ihorst, G.; Heinemann, V.; Hofheinz, R.; Moehler, M.; Buechler, P.; Kloeppel, G.; Röcken, C.; Bitzer, M.; Boeck, S.; et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br. J. Cancer 2012, 106, 1033–1038. [Google Scholar] [CrossRef]
- Li, L.; Luo, C.; Song, Z.; Reyes-Vargas, E.; Clayton, F.; Huang, J.; Jensen, P.; Chen, X. Association of anti-HER2 antibody with graphene oxide for curative treatment of osteosarcoma. Nanomedicine 2018, 14, 581–593. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Bay, A.E.P.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef]
- Luo, C.; Deng, Z.; Li, L.; Clayton, F.; Chen, A.L.; Wei, R.; Miles, R.; Stephens, D.M.; Glenn, M.; Wang, X.; et al. Association of rituximab with graphene oxide confers direct cytotoxicity for CD20-positive lymphoma cells. Oncotarget 2016, 7, 12806–12822. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fierens, K.; Zhang, Z.; Vanparijs, N.; Schuijs, M.J.; Van Steendam, K.; Feiner Gracia, N.; De Rycke, R.; De Beer, T.; De Beuckelaer, A.; et al. Spontaneous Protein Adsorption on Graphene Oxide Nanosheets Allowing Efficient Intracellular Vaccine Protein Delivery. ACS Appl. Mater. Interfaces 2016, 8, 1147–1155. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-Graphene Oxide for Cellular Imaging and Drug Delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef]
- Goenka, S.; Sant, V.; Sant, S. Graphene-based nanomaterials for drug delivery and tissue engineering. J. Control. Release 2014, 173, 75–88. [Google Scholar] [CrossRef]
- Shim, G.; Kim, M.G.; Park, J.Y.; Oh, Y.K. Graphene-based nanosheets for delivery of chemotherapeutics and biological drugs. Adv. Drug Deliv. Rev. 2016, 105, 205–227. [Google Scholar] [CrossRef]
- Yang, D.; Feng, L.; Dougherty, C.A.; Luker, K.E.; Chen, D.; Cauble, M.A.; Holl, M.M.B.; Luker, G.D.; Ross, B.D.; Liu, Z.; et al. In vivo targeting of metastatic breast cancer via tumor vasculature-specific nano-graphene oxide. Biomaterials 2016, 104, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.B.; Duffus, W.P.H.; Raff, M.C.; Petris, S.D. Redistribution and pinocytosis of lymphocyte surface immunoglobulin molecules induced by anti-immunoglobulin antibody. Nat. N. Biol. 1971, 233, 225–229. [Google Scholar] [CrossRef]
- Liu, S.; Zeng, T.H.; Hofmann, M.; Burcombe, E.; Wei, J.; Jiang, R.; Kong, J.; Chen, Y. Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: Membrane and oxidative stress. ACS Nano 2011, 5, 6971–6980. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [Green Version]
- Johnston, A.; Wang, Z. Necroptosis: MLKL Polymerization. J. Nat. Sci. 2018, 4, e513. [Google Scholar]
- Zhou, W.; Yuan, J. SnapShot: Necroptosis. Cell 2014, 158, 464. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Ali, M.; Mocarski, E.S. Proteasome inhibition blocks necroptosis by attenuating death complex aggregation. Cell Death Dis. 2018, 9, 346. [Google Scholar] [CrossRef]
- Esterberg, R.; Linbo, T.; Pickett, S.B.; Wu, P.; Ou, H.C.; Rubel, E.W.; Raible, D.W. Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J. Clin. Investig. 2016, 126, 3556–3566. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef]
- Koike, A.; Hanatani, M.; Fujimori, K. Pan-caspase inhibitors induce necroptosis via ROS-mediated activation of mixed lineage kinase domain-like protein and p38 in classically activated macrophages. Exp. Cell Res. 2019, 380, 171–179. [Google Scholar] [CrossRef]
- Shi, G.; Jia, P.; Chen, H.; Bao, L.; Feng, F.; Tang, H. Necroptosis occurs in osteoblasts during tumor necrosis factor-alpha stimulation and caspase-8 inhibition. Braz. J. Med. Biol. Res. 2018, 52, e7844. [Google Scholar] [CrossRef]
- Uluçkan, Ö.; Segaliny, A.; Botter, S.; Santiago, J.M.; Mutsaers, A.J. Preclinical mouse models of osteosarcoma. BoneKEy Rep. 2015, 4, 670. [Google Scholar] [CrossRef]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Hannes, S.; Abhari, B.A.; Fulda, S. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016, 380, 31–38. [Google Scholar] [CrossRef]
- Collart, M.A.; Baeuerle, P.; Vassalli, P. Regulation of tumor necrosis factor alpha transcription in macrophages: Involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol. Cell. Biol. 1990, 10, 1498–1506. [Google Scholar] [CrossRef]
- Kishore, R.; McMullen, M.R.; Cocuzzi, E.; Nagy, L.E. Lipopolysaccharide-mediated signal transduction: Stabilization of TNF-alpha mRNA contributes to increased lipopolysaccharide-stimulated TNF-alpha production by Kupffer cells after chronic ethanol feeding. Comp. Hepatol. 2004, 3, S31. [Google Scholar] [CrossRef]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef]
- Peng, C.; Cho, Y.Y.; Zhu, F.; Zhang, J.; Wen, W.; Xu, Y.; Yao, K.; Ma, W.Y.; Bode, A.M.; Dong, Z. Phosphorylation of caspase-8 (Thr-263) by ribosomal S6 kinase 2 (RSK2) mediates caspase-8 ubiquitination and stability. J. Biol. Chem. 2011, 286, 6946–6954. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Fiorillo, M.; Verre, A.F.; Iliut, M.; Peiris-Pagés, M.; Ozsvari, B.; Gandara, R.; Cappello, A.R.; Sotgia, F.; Vijayaraghavan, A.; Lisanti, M.P. Graphene oxide selectively targets cancer stem cells, across multiple tumor types: Implications for non-toxic cancer treatment, via “differentiation-based nano-therapy”. Oncotarget 2015, 6, 3553–3562. [Google Scholar] [CrossRef]
- Jin, G.; Liu, Y.; Xu, P. Induction of Necroptosis in Human Breast Cancer Drug-Resistant Cells by SMAC Analog LCL161 After Caspase Inhibition Requires RIP3. Pharmazie 2019, 74, 363–368. [Google Scholar]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and Cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Phillips, G.D.L.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.; Jensen, P.E.; Chen, X. Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies. Int. J. Mol. Sci. 2019, 20, 4360. https://doi.org/10.3390/ijms20184360
Xiao H, Jensen PE, Chen X. Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies. International Journal of Molecular Sciences. 2019; 20(18):4360. https://doi.org/10.3390/ijms20184360
Chicago/Turabian StyleXiao, Hongmei, Peter E. Jensen, and Xinjian Chen. 2019. "Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies" International Journal of Molecular Sciences 20, no. 18: 4360. https://doi.org/10.3390/ijms20184360
APA StyleXiao, H., Jensen, P. E., & Chen, X. (2019). Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies. International Journal of Molecular Sciences, 20(18), 4360. https://doi.org/10.3390/ijms20184360