Flow Cytometry Reveals the Nature of Oncotic Cells


Abstract
:1. Introduction
2. Results
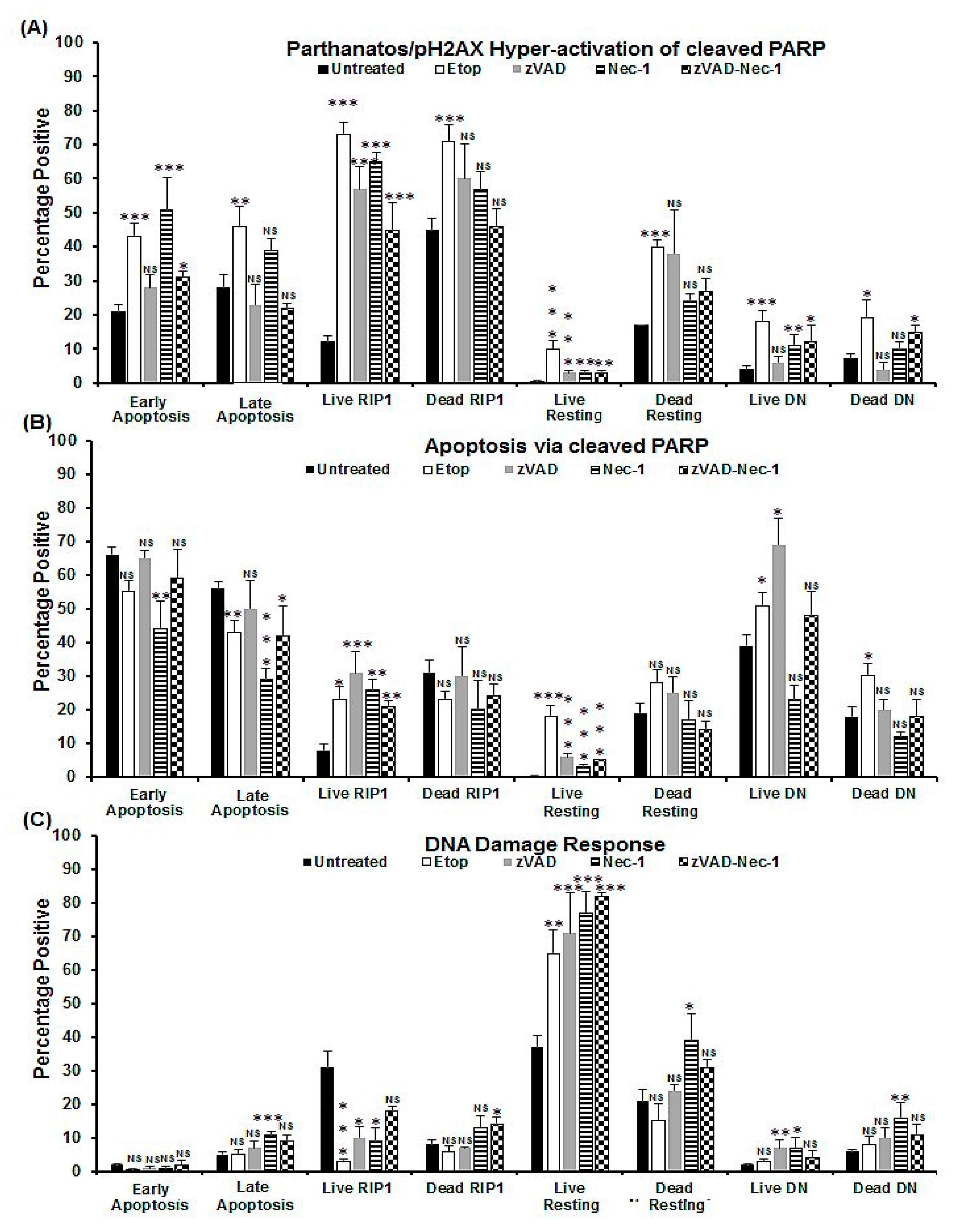
2.1. Induction of Oncosis
2.2. Induction of Apoptosis
2.3. Blockade of Caspases
2.4. Blockade with Necrostatin-1
2.5. Blockade with zVAD and Necrostatin-1
3. Discussion
4. Materials and Methods
4.1. Induction of Oncosis and Apoptosis
4.2. Flow Cytometry Assay
4.3. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, P.; Buja, L.M. Oncosis: An important non-apoptotic mode of cell death. Exp. Mol. Pathol. 2012, 93, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Joris, I. Apoptosis, Oncosis, and Necrosis. An Overview of Cell Death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Juan, G.; Li, X.; Gorczyca, W.; Murakami, T.; Traganos, F. Cytometry in Cell Necrobiology: Analysis of Apoptosis and Accidental Cell Death (Necrosis). Cytom. A 1997, 27, 1–20. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Trump, B.E.; Berezesky, I.K.; Chang, S.H.; Phelps, P.C. The Pathways of Cell Death: Oncosis, Apoptosis, and Necrosis. oxocological Pathol. 1997, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.M.; Xu, D.; Fergusson, M.M.; Combs, C.A.; Xu, Y.; Finkel, T. Regulation of cellular oncosis by uncoupling protein 2. J. Biol. Chem. 2002, 277, 27385–27392. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.; Martins, S. Real-time flow cytometry for the kinetic analysis of oncosis. Cytom. A 2011, 79, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Cytometry in cell necrobiology revisited. Recent advances and new vistas. Cytom. A 2010, 77A, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Lecoeur, H.; Prévost, M.C.; Gougeon, M.L. Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: A reconsideration of the specificity of the annexin V/propidium iodide assay. Cytom. A 2001, 44, 44–65. [Google Scholar] [CrossRef]
- Matteucci, C.; Grelli, S.; De Smaele, E.; Fontana, C.; Mastino, A. identification of nuclei from apoptotic necrotic and viable lymphoid cells by using multiparameter flow cytometry. Cytom. A 1999, 35, 145–153. [Google Scholar] [CrossRef]
- Lee, H.L.; Pike, R.; Chong, M.H.A.; Vossenkamper, A.; Warnes, G. Simultaneous flow cytometric immunophenotyping of necroptosis, apoptosis and RIP1-dependent apoptosis. Methods 2018, 134–135, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Vossenkamper, A.; Warnes, G. A flow cytometric immunophenotyping approach to the detection of regulated cell death processes. J. Immunol. Sci. 2018, 25, 6–12. [Google Scholar]
- Bergamaschi, D.; Vossenkamper, A.; Lee, W.Y.J.; Wang, P.; Bochukova, E.; Warnes, G. Simultaneous polychromatic flow cytometric detection of multiple forms of regulated cell death. Apoptosis 2019, 24, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; McQuade, T.; Zhang, H.; Zhang, J.; Chan, F.K. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS ONE 2011, 6, e23209. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, S.; Liang, H.; Yang, H.; Liu, L.; Zhou, K.; Xu, L.; Liu, J.; Yun, L.; Lai, B.; et al. Bcl-2 protects TK6 cells against hydroquinone-induced apoptosis through PARP-1 cytoplasm translocation and stabilizing mitochondrial membrane potential. Env. Mol. Mutagen. 2018, 59, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J.; Bourgeois, M.; Harbison, R.D. Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders. Cardiovasc. Toxicol. 2018, 18, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Yang, Y.; Zhang, Y.Y.; Xie, Z.; Zhao, X.Y.; Sun, Y.; Kong, W.J. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol. 2018, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Population | Phenotypic Markers |
|---|---|
| Live resting (or necroptotic) | Caspase-3–ve/Zombie NIR–ve/RIP3+ve |
| Live double negative (DN) | Caspase-3–ve/Zombie NIR–ve/RIP3–ve |
| Early apoptosis (EAPO) | Caspase-3+ve/Zombie NIR–ve/RIP3–ve |
| Live RIP1-dependent apoptosis (RIP1-APO) | Caspase-3+ve/Zombie NIR–ve/RIP3+ve |
| Late apoptosis (LAPO) | Caspase-3+ve/Zombie NIR+ve/RIP3–ve |
| Dead/necrotic/oncotic | Caspase-3–ve/Zombie NIR+ve |
| Dead resting (or necroptotic) | Caspase-3–ve/Zombie NIR+ve/RIP3+ve |
| Dead double negative (DN) | Caspase-3–ve/Zombie NIR+ve/RIP3–ve |
| Dead RIP1-dependent apoptosis (RIP1-APO) | Caspase-3+ve/Zombie NIR+ve/RIP3+ve |
| DNA damage response (DDR) | pH2AX+ve/Cleaved PARP–ve |
| Hyper-activation of cleaved PARP/parthanatos | pH2AX+ve/Cleaved PARP+ve |
| Cleaved PARP | pH2AX–ve/Cleaved PARP+ve |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vossenkamper, A.; Warnes, G. Flow Cytometry Reveals the Nature of Oncotic Cells. Int. J. Mol. Sci. 2019, 20, 4379. https://doi.org/10.3390/ijms20184379
Vossenkamper A, Warnes G. Flow Cytometry Reveals the Nature of Oncotic Cells. International Journal of Molecular Sciences. 2019; 20(18):4379. https://doi.org/10.3390/ijms20184379
Chicago/Turabian StyleVossenkamper, Anna, and Gary Warnes. 2019. "Flow Cytometry Reveals the Nature of Oncotic Cells" International Journal of Molecular Sciences 20, no. 18: 4379. https://doi.org/10.3390/ijms20184379
APA StyleVossenkamper, A., & Warnes, G. (2019). Flow Cytometry Reveals the Nature of Oncotic Cells. International Journal of Molecular Sciences, 20(18), 4379. https://doi.org/10.3390/ijms20184379