Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Hepatitis C Virus (HCV)
3. Nuclear Factor Erythroid 2 (NF-E2)-Related Factor 2 (Nrf2)
3.1. Domain Structure of Nrf2
3.2. Nrf2 Regulation
3.2.1. Canonical Nrf2 Activation
Keap1-Mediated Activation of Nrf2
βTrCP-Mediated Activation of Nrf2
Hrd1-Mediated Activation of Nrf2
3.2.2. Non-Canonical Nrf2 Activation
4. Autophagy
5. Reactive Oxygen Species (ROS) in HCV-Infected Cells
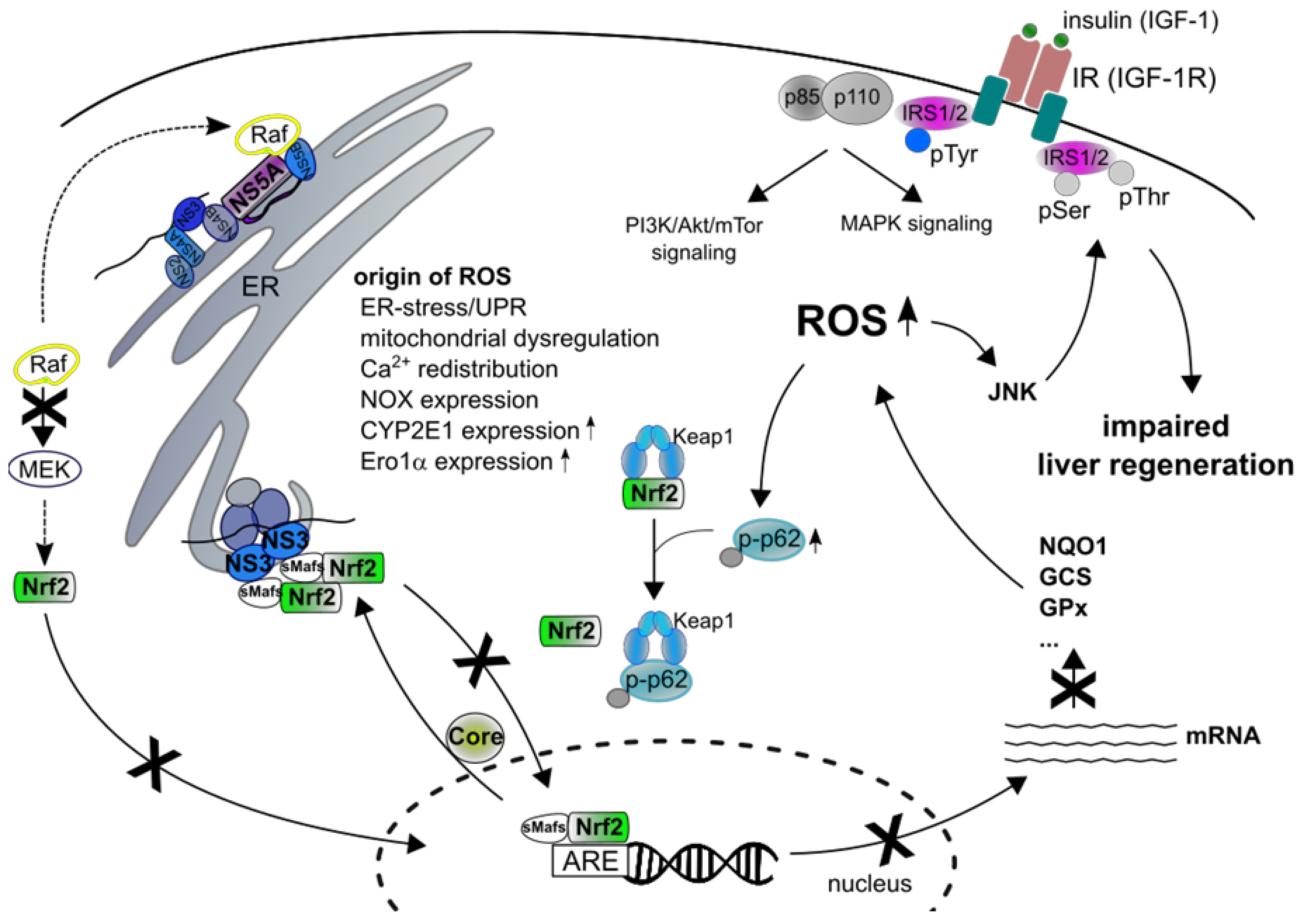
6. Interference of HCV with the Nrf2/Keap1-Signaling Pathway
7. Impact of ROS in HCV-Associated Pathogenesis
8. The Hepatitis B Virus (HBV)
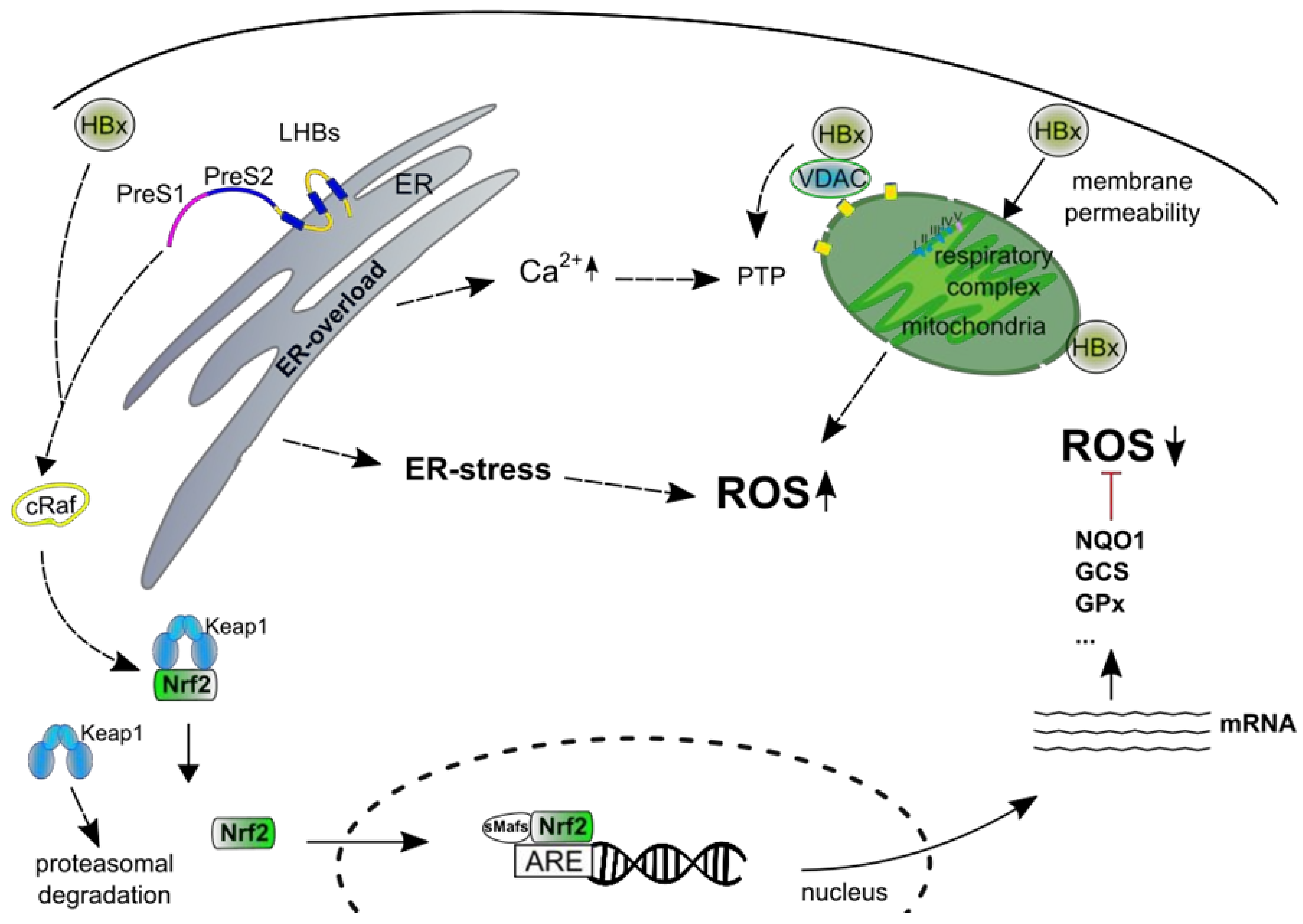
9. Generation of ROS in HBV-Replicating Cells
10. Interference of HBV with the Nrf2/Antioxidant Response Element (ARE) System
11. HBV Regulatory Proteins
12. Effect of ROS on the HBV Life-Cycle
13. ROS, Nrf2 and the Virus-Associated Pathogenesis
14. Liver Regeneration
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AhR | aryl hydrocarbon receptor |
| AKT | protein kinase B |
| AMPK | AMP-activated protein kinase |
| AMPKα | AMP-activated protein kinaseα |
| AP-1 | activating protein 1 |
| ARE | antioxidant-response element |
| ATF6 | activating transcription factor 6 |
| Atg | autophagy-regulated genes |
| Bach1/2 | BTB and CNC homology 1/2 |
| BCLN1 | Beclin1 |
| BRCA1 | breast cancer type 1 susceptibility protein |
| BTB | Broad-Complex, Tramtrack and Bric a brac |
| bZIP | basic-region leucine zipper |
| cat | catalase |
| CBP | CREB (cAMP response element-binding protein)-binding protein |
| CDH6 | chromo-ATPase/ helicase DNA-binding domain 6 |
| CHC | chronic hepatitis C |
| CK2 | casein kinase 2 |
| CNC | cap´n´collar |
| COX | cytochrome c oxidase |
| CTGF | connective tissue growth factor |
| CYP2E1 | cytochrome P450 2E1 |
| DAA | direct-acting antivirals |
| DFCP1 | double FYVE-containing protein 1 |
| DMV | double membrane vesicle |
| DNMTA3 | DNA methyltransferase |
| DPP3 | dipeptidyl peptidase III |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| ephx1 | epoxide hydrolase 1 |
| EpRE | electrophile-response element |
| ER | endoplasmatic reticulum |
| ERK | extracellular signal-regulated kinase |
| Ero1α | ER oxidoreductin 1α |
| ERQC | ER protein quality control |
| FGF | fibroblast growth factor |
| GABARAP | gamma-aminobutyric acid receptor-associated protein participate |
| GCLC | glutamate-cysteine ligase catalytic subunit |
| GLUT4 | glucose transporter 4 |
| GPx | glutathione peroxidase |
| GSH | glutathione |
| GSK3β | glycogen synthase kinase 3β |
| GSTM3 | glutathione-S-transferase M3 |
| GSTP1 | glutathione-S-transferase P1 |
| HBsAg | surface antigen |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HO-1 | heme oxygenase-1 |
| HSC | hepatic stellate cells |
| IGF-I | insulin-like growth factor I |
| IL-6 | interleukin 6 |
| IM | isolation membrane |
| IRE1 | inositol-required protein 1 |
| IRES | internal ribosomal entry site |
| JAK2 | Janus kinase 2 |
| JNK | c-Jun-N-terminal kinase |
| Keap1 | Kelch-like Ech-associated protein 1 |
| KIR | Keap1-interacting region |
| LC3 | microtubule-associated protein 1 light chain 3 |
| LD | lipid droplets |
| LHBs | large HBV surface protein |
| LIR | LC3 interacting region |
| LPS | lipopolysaccharide |
| LVP | lipoviroparticle |
| MAM | mitochondrial-associated membrane |
| MAPK | mitogen activated protein kinase |
| MCU | mitochondrial Ca2+-uniporter |
| MEF2D | myocyte enhancer factor 2d |
| MEOS | microsomal ethanol oxidizing enzymes |
| MHBs | middle surface |
| MPT | mitochondrial permeability transition |
| mPTP | mitochondrial permeability transition pore |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MVBs | multivesicular bodies |
| MW | membranous web |
| NAC | N-acetylcysteine |
| Neh | Nrf2-Ech homology |
| NF-κB | nuclear factor κB |
| NOX | NADPH oxidases |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| Nrf1 | NF-E2 related factor 1 |
| Nrf2 | nuclear factor erythroid 2 (NF-E2)-related factor 2 |
| NS | non-structural |
| ObR | obese receptor |
| OGG1 | 8-oxoguanine glycosylase 1 |
| PALB2 | partner and localizer for BRCA2 |
| PAS | phagophore assembly sites |
| PCK2 | phosphoenolpyruvate carboxykinase 2 |
| PDGF | platelet derived growth factor |
| PDI | protein disulfide isomerase |
| PE | phosphatidylethanolamine |
| PERK | protein kinase (PKR)-like ER kinase |
| PGC1α | peroxisome proliferator-activated receptor-gamma co-activator 1α |
| PI3K | phosphatidylinositol 3-kinase |
| PI3P | phosphatidylinositol-3-phosphate |
| PI4K4 | phosphatidylinositol 4-kinase IIIα |
| PKB | phosphatidylinositol 3-kinase (PI3K) kinase B |
| PKC | protein kinase C |
| PM | plasma membrane |
| PPARγ | peroxisome proliferator-activated receptor γ |
| ProTα | Prothymosin α |
| Rac | receptor-associated coactivator |
| ROS | reactive oxygen species |
| RXRα | retinoid X receptor α |
| SERCA | sarcoplasmic/endoplasmic reticulum calcium ATPase 2 |
| SHBs | proteinsmall surface protein |
| Skp1 | S-phase kinase-associated protein 1 |
| sMaf | v-maf avian musculoaponeurotic fibrosarcoma |
| SOCS3 | suppressor of cytokine signaling 3 |
| SOD2 | superoxide dismutase |
| SQSTM1 | sequestome 1 |
| β-TrCP | β-transducin repeat-containing protein |
| STAT3 | signal transducer and activator of transcription 3 |
| SUMO | small ubiquitin-like modifier |
| TAK1 | transforming growth factor β-activated kinase1 |
| TBK-1 | TANK-binding kinase 1 |
| tg | transgenic |
| TGF-β1 | transforming growth factor beta-1 |
| TNF-α | tumor necrosis factor α |
| TRAMP | transgenic adenocarcinoma of mouse prostate |
| TRIM16 | tripartite motif containing 16 |
| TSC | tuberous sclerosis complex |
| UBA | ubiquitin-associated |
| ULK1 | Unc-51-like kinase 1 |
| UPR | unfolded protein response |
| UTR | untranslated region |
| VDAC3 | voltage-dependent anion channel 3 |
| VLDL | very-low-density lipoprotein |
| Vps34 | vacuolar protein sorting 34 |
| WIPI | WD-repeat domain phosphoinositide-interacting |
| WTX | Wilms tumor gene in chromosome X |
| XBP1 | X box-binding protein 1 |
| XRE | xenobiotic response element |
| γ-GCS | γ-glutamylcysteine synthetase |
References
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015, 88, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerins, M.J.; Vashisht, A.A.; Liang, B.X.-T.; Duckworth, S.J.; Praslicka, B.J.; Wohlschlegel, J.A.; Ooi, A. Fumarate Mediates a Chronic Proliferative Signal in Fumarate Hydratase-Inactivated Cancer Cells by Increasing Transcription and Translation of Ferritin Genes. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Kuosmanen, S.M.; Kansanen, E.; Kaikkonen, M.U.; Sihvola, V.; Pulkkinen, K.; Jyrkkänen, H.-K.; Tuoresmäki, P.; Hartikainen, J.; Hippeläinen, M.; Kokki, H.; et al. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. 2018, 46, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Mimura, J.; Ozaki, T.; Itoh, K. Emerging Regulatory Role of Nrf2 in Iron, Heme, and Hemoglobin Metabolism in Physiology and Disease. Front. Vet. Sci. 2018, 5, 242. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Xu, W.; Teupser, D.; auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Werner, S. The cytoprotective Nrf2 transcription factor controls insulin receptor signaling in the regenerating liver. Cell Cycle 2008, 7, 874–878. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-F.; Kuo, H.-P.; Liu, M.; Chou, C.-K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.-T.; Huo, L.; Hsu, M.-C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell. 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.A.; Hyun, J.W. Oxidative Stress, Nrf2, and Epigenetic Modification Contribute to Anticancer Drug Resistance. Toxicol. Res. 2017, 33, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rojo de Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global hepatitis report. 2019. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 18 September 2019).
- Egerman, R.S. New Antiviral Agents for Treatment of Hepatitis C. Clin. Obstet. Gynecol. 2019. [Google Scholar] [CrossRef]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [Green Version]
- Bradley, D.; McCaustland, K.; Krawczynski, K.; Spelbring, J.; Humphrey, C.; Cook, E.H. Hepatitis C virus: Buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 1991, 34, 206–208. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Bréchot, C.; Paranhos-Baccalà, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.-S.; Brügger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Cosset, F.-L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.-Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Cortese, M.; Haselmann, U.; Tabata, K.; Romero-Brey, I.; Funaya, C.; Schieber, N.L.; Qiang, Y.; Bartenschlager, M.; Kallis, S.; et al. Spatiotemporal Coupling of the Hepatitis C Virus Replication Cycle by Creating a Lipid Droplet- Proximal Membranous Replication Compartment. Cell Rep. 2019, 27, 3602–3617.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeck, I.K.; Lee, J.-Y.; Tabata, K.; Romero-Brey, I.; Paul, D.; Schult, P.; Lohmann, V.; Kaderali, L.; Bartenschlager, R. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Falcón, V.; Acosta-Rivero, N.; González, S.; Dueñas-Carrera, S.; Martinez-Donato, G.; Menéndez, I.; Garateix, R.; Silva, J.A.; Acosta, E.; Kourı, J. Ultrastructural and biochemical basis for hepatitis C virus morphogenesis. Virus Genes 2017, 53, 151–164. [Google Scholar] [CrossRef]
- Elgner, F.; Ren, H.; Medvedev, R.; Ploen, D.; Himmelsbach, K.; Boller, K.; Hildt, E. The intra-cellular cholesterol transport inhibitor U18666A inhibits the exosome-dependent release of mature hepatitis C virus. J. Virol. 2016, 90, 11181–11196. [Google Scholar] [CrossRef]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-Mediated Intercellular Communication between Hepatitis C Virus-Infected Hepatocytes and Hepatic Stellate Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Yang, C.S.; Pickett, C.B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic. Biol. Med. 2004, 37, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharm.. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-K.; Kensler, T.W. Induction of 26S proteasome subunit PSMB5 by the bifunctional inducer 3-methylcholanthrene through the Nrf2-ARE, but not the AhR/Arnt-XRE, pathway. Biochem. Biophys. Res. Commun. 2006, 345, 1350–1357. [Google Scholar] [CrossRef]
- Chan, J.Y.; Han, X.L.; Kan, Y.W. Isolation of cDNA encoding the human NF-E2 protein. Proc. Natl. Acad. Sci. USA 1993, 90, 11366–11370. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Han, X.L.; Kan, Y.W. Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc. Natl. Acad. Sci. USA 1993, 90, 11371–11375. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Ito, E.; Toki, T.; Kogame, K.; Takahashi, S.; Igarashi, K.; Hayashi, N.; Yamamoto, M. Molecular cloning and functional characterization of a new Cap’n’ collar family transcription factor Nrf3. J. Biol. Chem. 1999, 274, 6443–6452. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, G.; Blank, V. NFE2L3 (NRF3): The Cinderella of the Cap’n’Collar transcription factors. Cell. Mol. Life Sci. 2011, 68, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, A.; Hoshino, H.; Madisen, L.; Yanai, N.; Obinata, M.; Karasuyama, H.; Hayashi, N.; Nakauchi, H.; Yamamoto, M.; Groudine, M.; et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3’ enhancer. EMBO J. 1998, 17, 5734–5743. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Tung, M.-C.; Lin, P.-L.; Wang, Y.-C.; He, T.-Y.; Lee, M.-C.; Yeh, S.D.; Chen, C.-Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget. 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Nagar, S.; Noveral, S.M.; Trudler, D.; Lopez, K.M.; McKercher, S.R.; Han, X.; Yates, J.R.; Piña-Crespo, J.C.; Nakanishi, N.; Satoh, T.; et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, E4048–E4056. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Rodriguez, O.C.; Nath, R.G.; Tilli, E.M.; Albanese, C.; Chung, F.-L.; Kwon, S.H.; Bae, I. Detoxification: A novel function of BRCA1 in tumor suppression? Toxicol. Sci. 2011, 122, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Werner, S. NRF2 and microRNAs: New but awaited relations. Biochem. Soc. Trans. 2015, 43, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Khor, T.O.; Cheung, K.-L.; Li, W.; Wu, T.-Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.-N. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.-N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; La Rojo de Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Liu, P.; Luo, G.; La Rojo de Vega, M.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lai, Q.; Chen, C.; Li, N.; Sun, F.; Huang, W.; Zhang, S.; Yu, Q.; Yang, P.; Xiong, F.; et al. Both conditional ablation and overexpression of E2 SUMO-conjugating enzyme (UBC9) in mouse pancreatic beta cells result in impaired beta cell function. Diabetologia. 2018, 61, 881–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Harder, B.; La Rojo de Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell. 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid. Redox Signal 2016, 25, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell. 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Ro, S.-H.; Semple, I.A.; Park, H.; Park, H.; Park, H.-W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef] [PubMed]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Søreng, K.; Neufeld, T.P.; Simonsen, A. Membrane Trafficking in Autophagy. Int. Rev. Cell Mol. Biol. 2018, 336, 1–92. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, M.; Karim, M.R.; Carpi, A.; Miotto, G. Nutrient control of macroautophagy in mammalian cells. Mol. Asp. Med. 2006, 27, 426–443. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell. 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell. 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebollero, E.; van der Vaart, A.; Zhao, M.; Rieter, E.; Klionsky, D.J.; Helms, J.B.; Reggiori, F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol. 2012, 22, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Mukhtarov, F.S.; Tunitskaya, V.L.; Jansons, J.; Isaguliants, M.G.; Kochetkov, S.N.; Ivanov, A.V. Analysis of the Domains of Hepatitis C Virus Core and NS5A Proteins that Activate the Nrf2/ARE Cascade. Acta Nat. 2016, 8, 123–127. [Google Scholar] [CrossRef]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.-P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.D.; Colberg-Poley, A.M. Access of viral proteins to mitochondria via mitochondria-associated membranes. Rev. Med. Virol. 2009, 19, 147–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yupeng, H.; Staschke Kirk, A.; Seng-Lai, T. HCV NS5A: A Multifunctional Regulator of Cellular Pathways and Virus Replication. Hepat. C Virus. Genom. Mol. Biol. 2006, 9, 1–22. [Google Scholar]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Seidel, J.; Biczysko, W.; Wysocki, J.; Spachacz, R.; Zabel, M. Intracellular localization of NS3 and C proteins in chronic hepatitis C. Liver Int. 2005, 25, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Jassey, A.; Liu, C.-H.; Changou, C.A.; Richardson, C.D.; Hsu, H.-Y.; Lin, L.-T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Bureau, C.; Bernad, J.; Chaouche, N.; Orfila, C.; Béraud, M.; Gonindard, C.; Alric, L.; Vinel, J.P.; Pipy, B. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 2001, 276, 23077–23083. [Google Scholar] [CrossRef]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. (Lond). 2000, 529 Pt 1, 37–47. [Google Scholar] [CrossRef]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.B.; Ott, M. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of Hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; de Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Dionisio, N.; Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez-Gallego, J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [Green Version]
- Siu, G.K.Y.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis C virus NS5A protein cooperates with phosphatidylinositol 4-kinase IIIα to induce mitochondrial fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1α regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Petrushanko, I.Y.; Ivanova, O.N.; Karpenko, I.L.; Alekseeva, E.; Sominskaya, I.; Makarov, A.A.; Bartosch, B.; Kochetkov, S.N.; et al. HCV core protein uses multiple mechanisms to induce oxidative stress in human hepatoma Huh7 cells. Viruses 2015, 7, 2745–2770. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.-W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar] [PubMed]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef] [PubMed]
- de Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A.V. Hepatitis C Virus NS5A Protein Triggers Oxidative Stress by Inducing NADPH Oxidases 1 and 4 and Cytochrome P450 2E1. Oxid. Med. Cell. Longev. 2016, 2016, 8341937. [Google Scholar] [CrossRef] [PubMed]
- Avadhani, N.G.; Sangar, M.C.; Bansal, S.; Bajpai, P. Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: The concept of chimeric signals. FEBS J. 2011, 278, 4218–4229. [Google Scholar] [CrossRef]
- Zeng, T.; Xie, K.-Q. Cytochrome P4502E1 Gene Polymorphisms and the Risks of Ethanol-Induced Health Problems in Alcoholics. Mol. Asp. Alcohol Nutr. 2016, 19, 231–245. [Google Scholar] [CrossRef]
- Wen, F.; Abdalla, M.Y.; Aloman, C.; Xiang, J.; Ahmad, I.M.; Walewski, J.; McCormick, M.L.; Brown, K.E.; Branch, A.D.; Spitz, D.R.; et al. Increased prooxidant production and enhanced susceptibility to glutathione depletion in HepG2 cells co-expressing HCV core protein and CYP2E1. J. Med. Virol. 2004, 72, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Ron, D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta. 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef]
- Ríos-Ocampo, W.A.; Navas, M.-C.; Faber, K.N.; Daemen, T.; Moshage, H. The cellular stress response in hepatitis C virus infection: A balancing act to promote viral persistence and host cell survival. Virus Res. 2019, 263, 1–8. [Google Scholar] [CrossRef]
- Paillet, J.; Kroemer, G. Autophagy represses hepatic carcinogenesis. Mol. Cell. Oncol. 2019, 6, 1573080. [Google Scholar] [CrossRef]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 9012580. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ou, J.-h.J. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sihvola, V.; Levonen, A.-L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Spengler, C.; Elgner, F.; Ren, H.; Bunten, S.; Hildt, E. HCV-induced oxidative stress by inhibition of Nrf2 triggers autophagy and favors release of viral particles. Free Radic. Biol. Med. 2017, 110, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Bürckstümmer, T.; Kriegs, M.; Lupberger, J.; Pauli, E.K.; Schmittel, S.; Hildt, E. Raf-1 kinase associates with Hepatitis C virus NS5A and regulates viral replication. FEBS Lett. 2006, 580, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Himmelsbach, K.; Kriegs, M.; Carvajal Yepes, M.; Hildt, E. Localization determines function: N-terminally truncated NS5A fragments accumulate in the nucleus and impair HCV replication. J. Hepatol. 2009, 50, 861–871. [Google Scholar] [CrossRef]
- Himmelsbach, K.; Sauter, D.; Baumert, T.F.; Ludwig, L.; Blum, H.E.; Hildt, E. New aspects of an anti-tumour drug: Sorafenib efficiently inhibits HCV replication. Gut 2009, 58, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.-A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar] [CrossRef] [PubMed]
- Blackham, S.; Baillie, A.; Al-Hababi, F.; Remlinger, K.; You, S.; Hamatake, R.; McGarvey, M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 2010, 84, 5404–5414. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Britigan, B.E.; Wen, F.; Icardi, M.; McCormick, M.L.; LaBrecque, D.R.; Voigt, M.; Brown, K.E.; Schmidt, W.N. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C core protein. J. Infect. Dis. 2004, 190, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol. 2010, 91, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Bao, H.; Ge, Y.; Tang, W.; Cheng, D.; Luo, K.; Gong, G.; Gong, R. Therapeutic targeting of GSK3beta enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C. Gut 2015, 64, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Ghaziani, T.; Shan, Y.; Lambrecht, R.W.; Donohue, S.E.; Pietschmann, T.; Bartenschlager, R.; Bonkovsky, H.L. HCV proteins increase expression of heme oxygenase-1 (HO-1) and decrease expression of Bach1 in human hepatoma cells. J. Hepatol. 2006, 45, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Bessa, S.S.E.-D.; Mohamed Ali, E.M.; Abd El-Wahab, A.E.-S.; Nor El-Din, S.A.E.-M. Heme oxygenase-1 mRNA expression in egyptian patients with chronic liver disease. Hepat. Mon. 2012, 12, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ocampo, W.A.; Daemen, T.; Buist-Homan, M.; Faber, K.N.; Navas, M.-C.; Moshage, H. Hepatitis C virus core or NS3/4A protein expression preconditions hepatocytes against oxidative stress and endoplasmic reticulum stress. Redox Rep. 2019, 24, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Anticoli, S.; Amatore, D.; Matarrese, P.; de Angelis, M.; Palamara, A.T.; Nencioni, L.; Ruggieri, A. Counteraction of HCV-Induced Oxidative Stress Concurs to Establish Chronic Infection in Liver Cell Cultures. Oxid. Med. Cell. Longev. 2019, 2019, 6452390. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Ni, H.-M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.-X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, R.E.; Wu, J. Mammalian target of rapamycin inhibition in hepatocellular carcinoma. World J. Hepatol. 2014, 6, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. The Lancet. 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Zhou, W.-C.; Zhang, Q.-B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Huh, C.-G.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef]
- Mead, J.E.; Fausto, N. Transforming growth factor alpha may be a physiological regulator of liver regeneration by means of an autocrine mechanism. Proc. Natl. Acad. Sci. USA 1989, 86, 1558–1562. [Google Scholar] [CrossRef]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef] [Green Version]
- Steiling, H.; Wüstefeld, T.; Bugnon, P.; Brauchle, M.; Fässler, R.; Teupser, D.; Thiery, J.; Gordon, J.I.; Trautwein, C.; Werner, S. Fibroblast growth factor receptor signalling is crucial for liver homeostasis and regeneration. Oncogene 2003, 22, 4380–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbois-Mouthon, C.; Wendum, D.; Cadoret, A.; Rey, C.; Leneuve, P.; Blaise, A.; Housset, C.; Tronche, F.; Le Bouc, Y.; Holzenberger, M. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J. 2006, 20, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Levéen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-β1: Role of TGF-β1 in HCV replication. Virology. 2011, 412, 284–296. [Google Scholar] [CrossRef]
- Wu, C.-F.; Lin, Y.-L.; Huang, Y.-T. Hepatitis C virus core protein stimulates fibrogenesis in hepatic stellate cells involving the obese receptor. J. Cell. Biochem. 2013, 114, 541–550. [Google Scholar] [CrossRef]
- Ming-Ju, H.; Yih-Shou, H.; Tzy-Yen, C.; Hui-Ling, C. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. J. Cell. Biochem. 2011, 112, 233–243. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Dotto, G.P. p21(WAF1/Cip1): More than a break to the cell cycle? Biochim. Biophys. Acta. 2000, 1471, M43–M56. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar] [PubMed]
- O’Reilly, M.A. Redox activation of p21Cip1/WAF1/Sdi1: A multifunctional regulator of cell survival and death. Antioxid. Redox Signal. 2005, 7, 108–118. [Google Scholar] [CrossRef]
- Wang, F.; Yoshida, I.; Takamatsu, M.; Ishido, S.; Fujita, T.; Oka, K.; Hotta, H. Complex formation between hepatitis C virus core protein and p21Waf1/Cip1/Sdi1. Biochem. Biophys. Res. Commun. 2000, 273, 479–484. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharm. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Leney, S.E.; Tavaré, J.M. The molecular basis of insulin-stimulated glucose uptake: Signalling, trafficking and potential drug targets. J. Endocrinol. 2009, 203, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zick, Y. Ser/Thr phosphorylation of IRS proteins: A molecular basis for insulin resistance. Sci. STKE 2005, 2005, pe4. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Ventura, J.-J.; Cogswell, P.; Flavell, R.A.; Baldwin, A.S.; Davis, R.J. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralj, D.; Virović Jukić, L.; Stojsavljević, S.; Duvnjak, M.; Smolić, M.; Čurčić, I.B. Hepatitis C Virus, Insulin Resistance, and Steatosis. J. Clin. Transl. Hepatol. 2016, 4, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Tsukamoto, K.; Kimura, S.; Moriya, K.; Koike, K. Hepatitis C virus infection and diabetes: Direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004, 126, 840–848. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Yoshida, T.; Harada, M.; Hisamoto, T.; Nagao, Y.; Ide, T.; Taniguchi, E.; Kumemura, H.; Hanada, S.; Maeyama, M.; et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 2004, 165, 1499–1508. [Google Scholar] [CrossRef]
- Banerjee, S.; Saito, K.; Ait-Goughoulte, M.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 2008, 82, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Meyer, K.; Mazumdar, B.; Ray, R.B.; Ray, R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 2010, 84, 5936–5946. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumashiro, N.; Tamura, Y.; Uchida, T.; Ogihara, T.; Fujitani, Y.; Hirose, T.; Mochizuki, H.; Kawamori, R.; Watada, H. Impact of oxidative stress and peroxisome proliferator-activated receptor gamma coactivator-1alpha in hepatic insulin resistance. Diabetes 2008, 57, 2083–2091. [Google Scholar] [CrossRef]
- Shlomai, A.; Rechtman, M.M.; Burdelova, E.O.; Zilberberg, A.; Hoffman, S.; Solar, I.; Fishman, S.; Halpern, Z.; Sklan, E.H. The metabolic regulator PGC-1α links hepatitis C virus infection to hepatic insulin resistance. J. Hepatol. 2012, 57, 867–873. [Google Scholar] [CrossRef]
- Pazienza, V.; Clément, S.; Pugnale, P.; Conzelman, S.; Foti, M.; Mangia, A.; Negro, F. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology 2007, 45, 1164–1171. [Google Scholar] [CrossRef]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Investig. 2008, 118, 683–694. [Google Scholar] [CrossRef]
- Toshima, T.; Shirabe, K.; Fukuhara, T.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Okano, S.; Maehara, Y. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology 2014, 60, 290–300. [Google Scholar] [CrossRef]
- Köhler, U.A.; Kurinna, S.; Schwitter, D.; Marti, A.; Schäfer, M.; Hellerbrand, C.; Speicher, T.; Werner, S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 2014, 60, 670–678. [Google Scholar] [CrossRef]
- Haga, S.; Ozawa, T.; Yamada, Y.; Morita, N.; Nagashima, I.; Inoue, H.; Inaba, Y.; Noda, N.; Abe, R.; Umezawa, K.; et al. p62/SQSTM1 plays a protective role in oxidative injury of steatotic liver in a mouse hepatectomy model. Antioxid. Redox Signal. 2014, 21, 2515–2530. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, W.H. Medical virology of hepatitis B: How it began and where we are now. Virol. J. 2013, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [PubMed]
- Bruss, V. Hepatitis B virus morphogenesis. World J. Gastroenterol. 2007, 13, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patient, R.; Hourioux, C.; Sizaret, P.-Y.; Trassard, S.; Sureau, C.; Roingeard, P. Hepatitis B virus subviral envelope particle morphogenesis and intracellular trafficking. J. Virol. 2007, 81, 3842–3851. [Google Scholar] [CrossRef]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2015, 90, 3330–3341. [Google Scholar] [CrossRef]
- Levrero, M.; Testoni, B.; Zoulim, F. HBV cure: Why, how, when? Curr. Opin. Virol. 2016, 18, 135–143. [Google Scholar] [CrossRef]
- Chisari, F.V.; Isogawa, M.; Wieland, S.F. Pathogenesis of hepatitis B virus infection. Pathol. Biol. 2010, 58, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Bertoletti, A. Immunological aspects of antiviral therapy of chronic hepatitis B virus and hepatitis C virus infections. Hepatology 2015, 61, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.; Wong, R.J.; Gish, R.G. Global Perspective on Hepatitis B Virus Infections in the Era of Effective Vaccines. Clin. Liver Dis. 2019, 23, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Dastgerdi, E.; Schwartz, R.E.; Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McClain, S.L.; Clippinger, A.J.; Lizzano, R.; Bouchard, M.J. Hepatitis B virus replication is associated with an HBx-dependent mitochondrion-regulated increase in cytosolic calcium levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.-Y.; Zheng, B.-Y.; Fang, X.-F.; Li, D.; Huang, Y.-H.; Chen, Z.-X.; Zhou, L.-Y.; Wang, X.-Z. HBx co-localizes with COXIII in HL-7702 cells to upregulate mitochondrial function and ROS generation. Oncol. Rep. 2015, 33, 2461–2467. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Kwon, S.-H.; Cho, H.; Kim, S.; Lee, S.; Ryu, W.-S.; Cho, H. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J. Mol. Med. 2010, 88, 359–369. [Google Scholar] [CrossRef]
- Li, S.K.; Ho, S.F.; Tsui, K.W.; Fung, K.P.; Waye, M.Y.M. Identification of functionally important amino acid residues in the mitochondria targeting sequence of hepatitis B virus X protein. Virology 2008, 381, 81–88. [Google Scholar] [CrossRef]
- Jung, S.-Y.; Kim, Y.-J. C-terminal region of HBx is crucial for mitochondrial DNA damage. Cancer Lett. 2013, 331, 76–83. [Google Scholar] [CrossRef]
- Lee, H.-R.; Cho, Y.Y.; Lee, G.Y.; You, D.-G.; Yoo, Y.D.; Kim, Y.J. A direct role for hepatitis B virus X protein in inducing mitochondrial membrane permeabilization. J. Viral Hepat. 2018, 25, 412–420. [Google Scholar] [CrossRef]
- You, D.-G.; Cho, Y.Y.; Lee, H.-R.; Lee, J.-H.; Yu, S.J.; Yoon, J.-H.; Yoo, Y.D.; Kim, Y.J.; Lee, G.Y. Hepatitis B virus X protein induces size-selective membrane permeabilization through interaction with cardiolipin. Biochim. Biophys. Acta Biomembr. 2019, 1861, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ding, J.; Chen, Z.; Chen, Y.; Lin, N.; Chen, F.; Wang, X. Accurately mapping the location of the binding site for the interaction between hepatitis B virus X protein and cytochrome c oxidase III. Int. J. Mol. Med. 2015, 35, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Reina, S.; Guarino, F.; Magrì, A.; de Pinto, V. VDAC3 As a Potential Marker of Mitochondrial Status Is Involved in Cancer and Pathology. Front. Oncol. 2016, 6, 264. [Google Scholar] [CrossRef] [Green Version]
- de Pinto, V.; Guarino, F.; Guarnera, A.; Messina, A.; Reina, S.; Tomasello, F.M.; Palermo, V.; Mazzoni, C. Characterization of human VDAC isoforms: A peculiar function for VDAC3? Biochim. Biophys. Acta 2010, 1797, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Bouchard, M.J. The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J. Virol. 2012, 86, 313–327. [Google Scholar] [CrossRef]
- Casciano, J.C.; Bouchard, M.J. Hepatitis B virus X protein modulates cytosolic Ca2+ signaling in primary human hepatocytes. Virus Res. 2018, 246, 23–27. [Google Scholar] [CrossRef]
- Chisari, F.V.; Pinkert, C.A.; Milich, D.R.; Filippi, P.; McLachlan, A.; Palmiter, R.D.; Brinster, R.L. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science 1985, 230, 1157–1160. [Google Scholar] [CrossRef]
- Chisari, F.V.; Filippi, P.; McLachlan, A.; Milich, D.R.; Riggs, M.; Lee, S.; Palmiter, R.D.; Pinkert, C.A.; Brinster, R.L. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J. Virol. 1986, 60, 880–887. [Google Scholar] [Green Version]
- Choi, Y.-M.; Lee, S.-Y.; Kim, B.-J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, E597. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Caselmann, W.H.; Schlüter, V.; Schreck, R.; Hofschneider, P.H.; Baeuerle, P.A. Hepatitis B virus transactivator MHBst: Activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992, 11, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-F. Hepatitis B virus pre-S/S variants in liver diseases. World J. Gastroenterol. 2018, 24, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Caselmann, W.H.; Renner, M.; Schlüter, V.; Hofschneider, P.H.; Koshy, R.; Meyer, M. The hepatitis B virus MHBst167 protein is a pleiotropic transactivator mediating its effect via ubiquitous cellular transcription factors. J. Gen. Virol. 1997, 78 Pt 6, 1487–1495. [Google Scholar] [CrossRef]
- Hildt, E.; Urban, S.; Lauer, U.; Hofschneider, P.H.; Kekulé, A.S. ER-localization and functional expression of the HBV transactivator MHBst. Oncogene 1993, 8, 3359–3367. [Google Scholar] [PubMed]
- Hildt, E.; Urban, S.; Hofschneider, P.H. Characterization of essential domains for the functionality of the MHBst transcriptional activator and identification of a minimal MHBst activator. Oncogene 1995, 11, 2055–2066. [Google Scholar]
- Hildt, E.; Saher, G.; Bruss, V.; Hofschneider, P.H. The hepatitis B virus large surface protein (LHBs) is a transcriptional activator. Virology 1996, 225, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Bruss, V.; Lu, X.; Thomssen, R.; Gerlich, W.H. Post-translational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J. 1994, 13, 2273–2279. [Google Scholar] [CrossRef]
- Ostapchuk, P.; Hearing, P.; Ganem, D. A dramatic shift in the transmembrane topology of a viral envelope glycoprotein accompanies hepatitis B viral morphogenesis. EMBO J. 1994, 13, 1048–1057. [Google Scholar] [CrossRef]
- Prange, R.; Streeck, R.E. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J. 1995, 14, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Stöckl, L.; Berting, A.; Malkowski, B.; Foerste, R.; Hofschneider, P.H.; Hildt, E. Integrity of c-Raf-1/MEK signal transduction cascade is essential for hepatitis B virus gene expression. Oncogene 2003, 22, 2604–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayoub, R.; Vogel, A.; Schuett, J.; Lupke, M.; Spieker, S.M.; Kettern, N.; Hildt, E.; Melter, M.; Weiss, T.S. Nrf2 activates augmenter of liver regeneration (ALR) via antioxidant response element and links oxidative stress to liver regeneration. Mol. Med. 2013, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Morales-González, J.A.; Madrigal-Santillán, E.; Morales-González, Á.; Bautista, M.; Gayosso-Islas, E.; Sánchez-Moreno, C. What is Known Regarding the Participation of Factor Nrf-2 in Liver Regeneration? Cells 2015, 4, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, S.R.; Medvedev, R.; Heinrich, T.; Büchner, S.M.; Kettern, N.; Hildt, E. Hepatitis B virus inhibits insulin receptor signaling and impairs liver regeneration via intracellular retention of the insulin receptor. Cell. Mol. Life Sci. 2016, 73, 4121–4140. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage. J. Appl. Toxicol. 2019, 1–18. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Ki, S.H.; Brooks, C.; Kim, S.G. Role of hepatitis B virus X repression of C/EBPbeta activity in the down-regulation of glutathione S-transferase A2 gene: Implications in other phase II detoxifying enzyme expression. Xenobiotica 2009, 39, 182–192. [Google Scholar] [CrossRef]
- Niu, D.; Zhang, J.; Ren, Y.; Feng, H.; Chen, W.N. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol. 2009, 3, 67–76. [Google Scholar] [CrossRef]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef]
- Cheng, M.-L.; Lu, Y.-F.; Chen, H.; Shen, Z.-Y.; Liu, J. Liver expression of Nrf2-related genes in different liver diseases. HBPD INT 2015, 14, 485–491. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, K.-H.; Akhras, S.; Himmelsbach, K.; Hassemer, M.; Finkernagel, M.; Carra, G.; Nuebling, M.; Chudy, M.; Niekamp, H.; Glebe, D.; et al. Intracellular accumulation of subviral HBsAg particles and diminished Nrf2 activation in HBV genotype G expressing cells lead to an increased ROI level. J. Hepatol. 2015, 62, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Wang, D.; Peng, X.-E.; Chen, Y.-L.; Zheng, D.-L.; Chen, W.-N.; Lin, X. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic. Biol. Med. 2013, 65, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Liu, W.; He, Y.; Wu, Y.-L.; Chen, W.-N.; Lin, X.-J.; Lin, X. Hepatitis B Virus X Protein Increases 8-Oxo-7,8-Dihydro-2’-Deoxyguanosine (8-Oxodg) Level via Repressing MTH1/ MTH2 Expression in Hepatocytes. Cell. Physiol. Biochem. 2018, 51, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Song, X.; Li, Y.; Tan, D.; Liu, G. Hepatitis B virus X protein upregulates DNA methyltransferase 3A/3B and enhances SOCS-1CpG island methylation. Mol. Med. Rep. 2016, 13, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Lee, Y.-g.; Bae, J.-B.; Choi, J.K.; Tayama, C.; Hata, K.; Yun, Y.; Seong, J.-K.; Kim, Y.-J. HBx induces hypomethylation of distal intragenic CpG islands required for active expression of developmental regulators. Proc. Natl. Acad. Sci. USA 2014, 111, 9555–9560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Zou, Z.-Q.; Wang, L.-Y.; Gao, S.; Fan, Y.-C.; Long, B.; Guo, Y.-M.; Xu, A.-L.; Han, J.; Li, T.; et al. Methylation of the glutathione-S-transferase M3 gene promoter is associated with oxidative stress in acute-on-chronic hepatitis B liver failure. Tohoku J. Exp. Med. 2012, 228, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Shaban, N.Z.; Salem, H.H.; Elsadany, M.A.; Ali, B.A.; Hassona, E.M.; Mogahed, F.A. Distribution of Glutathione S-Transferase Omega Gene Polymorphism with Different Stages of HBV Infection Including Hepatocellular Carcinoma in the Egyptian Population. Asian Pac. J. Cancer Prev. 2016, 17, 2145–2150. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, J.; Lin, C.; Wang, H.; Jiang, Y.; Wang, J.; Yang, P.; He, F. Peroxiredoxin 2: A potential biomarker for early diagnosis of hepatitis B virus related liver fibrosis identified by proteomic analysis of the plasma. BMC Gastroenterol. 2010, 10, 115. [Google Scholar] [CrossRef]
- Katrinli, S.; Ozdil, K.; Sahin, A.; Ozturk, O.; Kir, G.; Baykal, A.T.; Akgun, E.; Sarac, O.S.; Sokmen, M.; Doğanay, H.L.; et al. Proteomic profiling of HBV infected liver biopsies with different fibrotic stages. Proteome Sci. 2016, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Seo, H.W.; Jung, G. Reactive oxygen species promote heat shock protein 90-mediated HBV capsid assembly. Biochem. Biophys. Res. Commun. 2015, 457, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Hildt, E.; Hofschneider, P.H. Anti-hepatitis B virus activity of N-acetyl-L-cysteine (NAC): New aspects of a well-established drug. Antiviral Res. 1996, 32, 43–53. [Google Scholar] [CrossRef]
- Ren, J.-H.; Chen, X.; Zhou, L.; Tao, N.-N.; Zhou, H.-Z.; Liu, B.; Li, W.-Y.; Huang, A.-L.; Chen, J. Protective Role of Sirtuin3 (SIRT3) in Oxidative Stress Mediated by Hepatitis B Virus X Protein Expression. PLoS ONE 2016, 11, e0150961. [Google Scholar] [CrossRef]
- Kitada, T.; Seki, S.; Iwai, S.; Yamada, T.; Sakaguchi, H.; Wakasa, K. In situ detection of oxidative DNA damage, 8-hydroxydeoxyguanosine, in chronic human liver disease. J. Hepatol. 2001, 35, 613–618. [Google Scholar] [CrossRef]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef]
- Tasdelen Fisgin, N.; Aydin, B.K.; Sarikaya, H.; Tanyel, E.; Esen, S.; Sunbul, M.; Leblebicioglu, H. Oxidative stress and antioxidant defense in patients with chronic hepatitis B. Clin. Lab. 2012, 58, 273–280. [Google Scholar]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Lucito, R.; Schneider, R.J. Hepatitis B virus X protein activates transcription factor NF-kappa B without a requirement for protein kinase C. J. Virol. 1992, 66, 983–991. [Google Scholar] [Green Version]
- Lauer, U.; Weiss, L.; Lipp, M.; Hofschneider, P.H.; Kekulé, A.S. The hepatitis B virus preS2/St transactivator utilizes AP-1 and other transcription factors for transactivation. Hepatology 1994, 19, 23–31. [Google Scholar] [CrossRef]
- Shokri, S.; Mahmoudvand, S.; Taherkhani, R.; Farshadpour, F.; Jalalian, F.A. Complexity on modulation of NF-κB pathways by hepatitis B and C: A double-edged sword in hepatocarcinogenesis. J. Cell. Physiol. 2019, 234, 14734–14742. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quétier, I.; Brezillon, N.; Duriez, M.; Massinet, H.; Giang, E.; Ahodantin, J.; Lamant, C.; Brunelle, M.-N.; Soussan, P.; Kremsdorf, D. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J. Hepatol. 2013, 59, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.X.M.; Wong, S.H.; Chan, M.T.V.; Yu, L.; Yu, S.S.B.; Wu, F.; Xiao, Z.; Wang, X.; Zhang, L.; Cheng, A.S.L.; et al. Autophagy Mediates HBx-Induced Nuclear Factor-κB Activation and Release of IL-6, IL-8, and CXCL2 in Hepatocytes. J. Cell. Physiol. 2015, 230, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; You, H.; Zhao, J.; Liu, W.; Hu, L.; Luo, W.; Hu, W.; Tang, R.; Zheng, K. The enhanced expression of death receptor 5 (DR5) mediated by HBV X protein through NF-kappaB pathway is associated with cell apoptosis induced by (TNF-α related apoptosis inducing ligand) TRAIL in hepatoma cells. Virol. J. 2015, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-H.; Su, I.-J.; Wang, H.-C.; Chang, W.-W.; Lei, H.-Y.; Lai, M.-D.; Chang, W.-T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef] [Green Version]
- Gwak, G.-Y.; Lee, D.H.; Moon, T.G.; Choi, M.S.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Park, C.K.; Joh, J.-W.; Yoo, B.C. The correlation of hepatitis B virus pre-S mutation with cellular oxidative DNA damage in hepatocellular carcinoma. Hepatogastroenterology 2008, 55, 2028–2032. [Google Scholar] [PubMed]
- Cheng, T.-L.; Chen, P.-S.; Li, R.-H.; Yuan, S.-S.; Su, I.-J.; Hung, J.-H. Induction of apurinic endonuclease 1 overexpression by endoplasmic reticulum stress in hepatoma cells. Int. J. Mol. Sci. 2014, 15, 12442–12457. [Google Scholar] [CrossRef]
- Hsieh, Y.-H.; Chang, Y.-Y.; Su, I.-J.; Yen, C.-J.; Liu, Y.-R.; Liu, R.-J.; Hsieh, W.-C.; Tsai, H.-W.; Wang, L.H.-C.; Huang, W. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J. Pathol. 2015, 236, 337–347. [Google Scholar] [CrossRef]
- Yen, T.T.-C.; Yang, A.; Chiu, W.-T.; Li, T.-N.; Wang, L.-H.; Wu, Y.-H.; Wang, H.-C.; Chen, L.; Wang, W.-C.; Huang, W.; et al. Hepatitis B virus PreS2-mutant large surface antigen activates store-operated calcium entry and promotes chromosome instability. Oncotarget 2016, 7, 23346–23360. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, H.-S.; Ji, J.-H.; Cho, M.-Y.; Yoo, Y.-S.; Park, Y.-Y.; Cha, H.-J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays cell cycle progression. J. Gen. Virology 2015, 96, 2242–2251. [Google Scholar] [CrossRef]
- Pitot, H.C.; Sirica, A.E. The stages of initiation and promotion in hepatocarcinogenesis. Biochim. Biophys. Acta 1980, 605, 191–215. [Google Scholar] [CrossRef]
- Potter, V.R. Initiation and promotion in cancer formation: The importance of studies on intercellular communication. Yale J. Biol. Med. 1980, 53, 367–384. [Google Scholar] [PubMed]
- Lupberger, J.; Hildt, E. Hepatitis B virus-induced oncogenesis. World J. Gastroenterol. 2007, 13, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivière, L.; Ducroux, A.; Buendia, M.A. The oncogenic role of hepatitis B virus. Recent Results Cancer Res. 2014, 193, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, H.; Lin, J.; Ye, D. Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. J. Cancer 2018, 9, 1182–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley, R.P.; Hwang, L.Y.; Lin, C.C.; Chien, C.S. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. A prospective study of 22 707 men in Taiwan. Lancet 1981, 2, 1129–1133. [Google Scholar] [CrossRef]
- Koshiol, J.; Liu, Z.; O’Brien, T.R.; Hildesheim, A. Beasley’s 1981 paper: The power of a well-designed cohort study to drive liver cancer research and prevention. Cancer Epidemiol. 2018, 53, 195–199. [Google Scholar] [CrossRef]
- Wei, Y.; Etiemble, J.; Fourel, G.; Vitvitski-Trepo, L.; Buendia, M.A. Hepadna virus integration generates virus-cell cotranscripts carrying 3’ truncated X genes in human and woodchuck liver tumors. J. Med. Virol. 1995, 45, 82–90. [Google Scholar] [CrossRef]
- Flajolet, M.; Gegonne, A.; Ghysdael, J.; Tiollais, P.; Buendia, M.A.; Fourel, G. Cellular and viral trans-acting factors modulate N-myc2 promoter activity in woodchuck liver tumors. Oncogene 1997, 15, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Podlaha, O.; Wu, G.; Downie, B.; Ramamurthy, R.; Gaggar, A.; Subramanian, M.; Ye, Z.; Jiang, Z. Genomic modeling of hepatitis B virus integration frequency in the human genome. PLoS ONE 2019, 14, e0220376. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.-H.; Cheng, S.C.Y.; Lung-Yi Mak, L.; Wai-Pan To, E.; Cheuk-Lam Lo, R.; Cheung, T.-T.; Seto, W.-K.; Fung, J.; Man, K.; Lai, C.-L.; et al. Among Patients with Undetectable Hepatitis B Surface Antigen and Hepatocellular Carcinoma, a High Proportion Has Integration of HBV DNA into Hepatocyte DNA and No Cirrhosis. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.R.; Sterczer, A.; Toshkov, I.A.; Yeager, A.E.; Korba, B.E.; Cote, P.J.; Buendia, M.-A.; Gerin, J.L.; Tennant, B.C. Integration of woodchuck hepatitis and N-myc rearrangement determine size and histologic grade of hepatic tumors. Hepatology 2004, 39, 1008–1016. [Google Scholar] [CrossRef]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.-M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Hoffmann, J.; Boehm, C.; Himmelsbach, K.; Donnerhak, C.; Roettger, H.; Weiss, T.S.; Ploen, D.; Hildt, E. Identification of α-taxilin as an essential factor for the life cycle of hepatitis B virus. J. Hepatol. 2013, 59, 934–941. [Google Scholar] [CrossRef]
- Nogami, S.; Satoh, S.; Nakano, M.; Terano, A.; Shirataki, H. Interaction of taxilin with syntaxin which does not form the SNARE complex. Biochem. Biophys. Res. Commun. 2003, 311, 797–802. [Google Scholar] [CrossRef]
- Nogami, S.; Satoh, S.; Nakano, M.; Shimizu, H.; Fukushima, H.; Maruyama, A.; Terano, A.; Shirataki, H. Taxilin; a novel syntaxin-binding protein that is involved in Ca2+-dependent exocytosis in neuroendocrine cells. Genes Cells 2003, 8, 17–28. [Google Scholar] [CrossRef]
- Dandri, M.; Schirmacher, P.; Rogler, C.E. Woodchuck hepatitis virus X protein is present in chronically infected woodchuck liver and woodchuck hepatocellular carcinomas which are permissive for viral replication. J. Virol. 1996, 70, 5246–5254. [Google Scholar] [PubMed]
- Hafner, A.; Brandenburg, B.; Hildt, E. Reconstitution of gene expression from a regulatory-protein-deficient hepatitis B virus genome by cell-permeable HBx protein. EMBO Rep. 2003, 4, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 20, 4659. https://doi.org/10.3390/ijms20184659
Bender D, Hildt E. Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. International Journal of Molecular Sciences. 2019; 20(18):4659. https://doi.org/10.3390/ijms20184659
Chicago/Turabian StyleBender, Daniela, and Eberhard Hildt. 2019. "Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis" International Journal of Molecular Sciences 20, no. 18: 4659. https://doi.org/10.3390/ijms20184659
APA StyleBender, D., & Hildt, E. (2019). Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. International Journal of Molecular Sciences, 20(18), 4659. https://doi.org/10.3390/ijms20184659