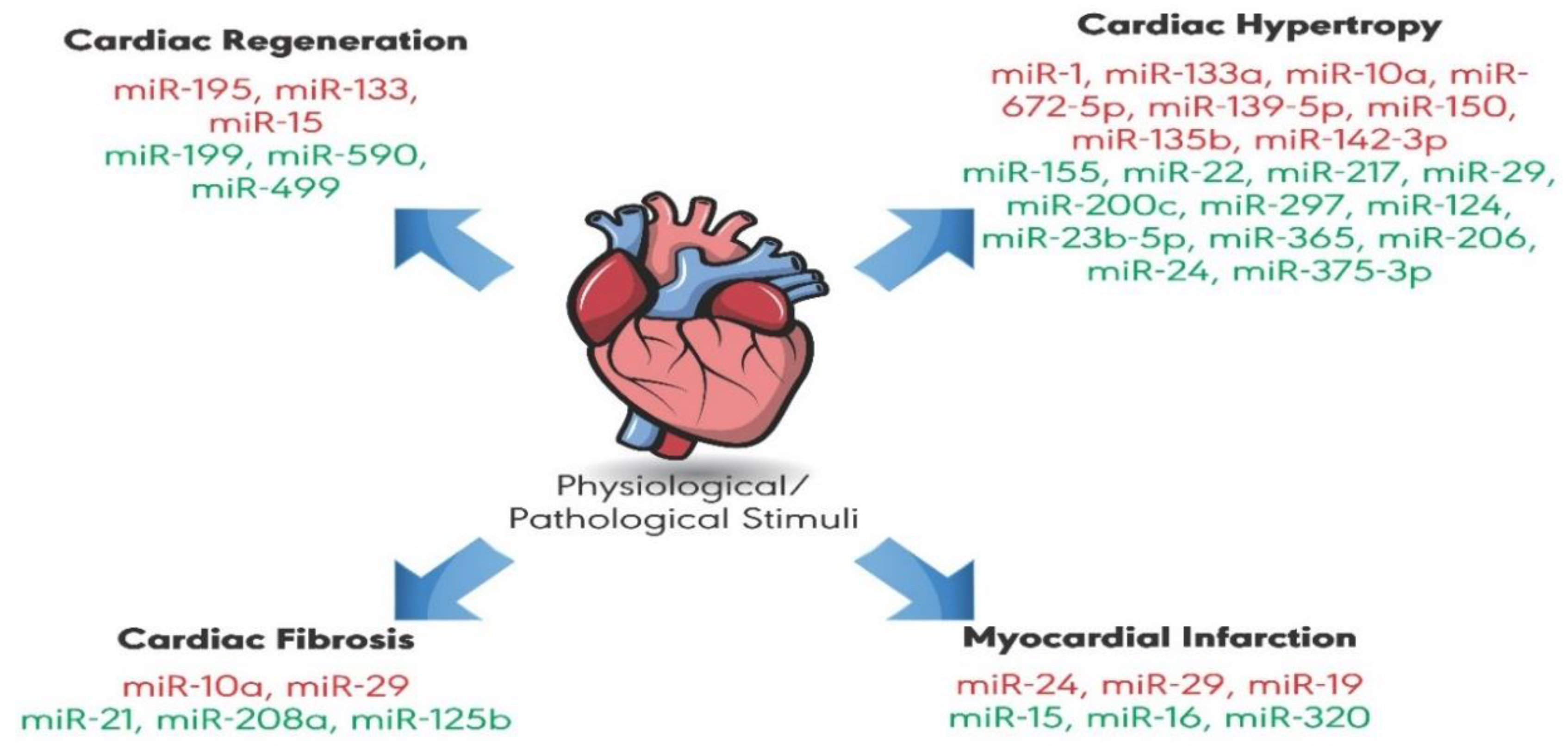
MicroRNAs in Cardiac Hypertrophy


 ,
,
Abstract
:1. Introduction
2. MicroRNAs (miRNAs)
3. MicroRNAs that Attenuate Cardiac Hypertrophy
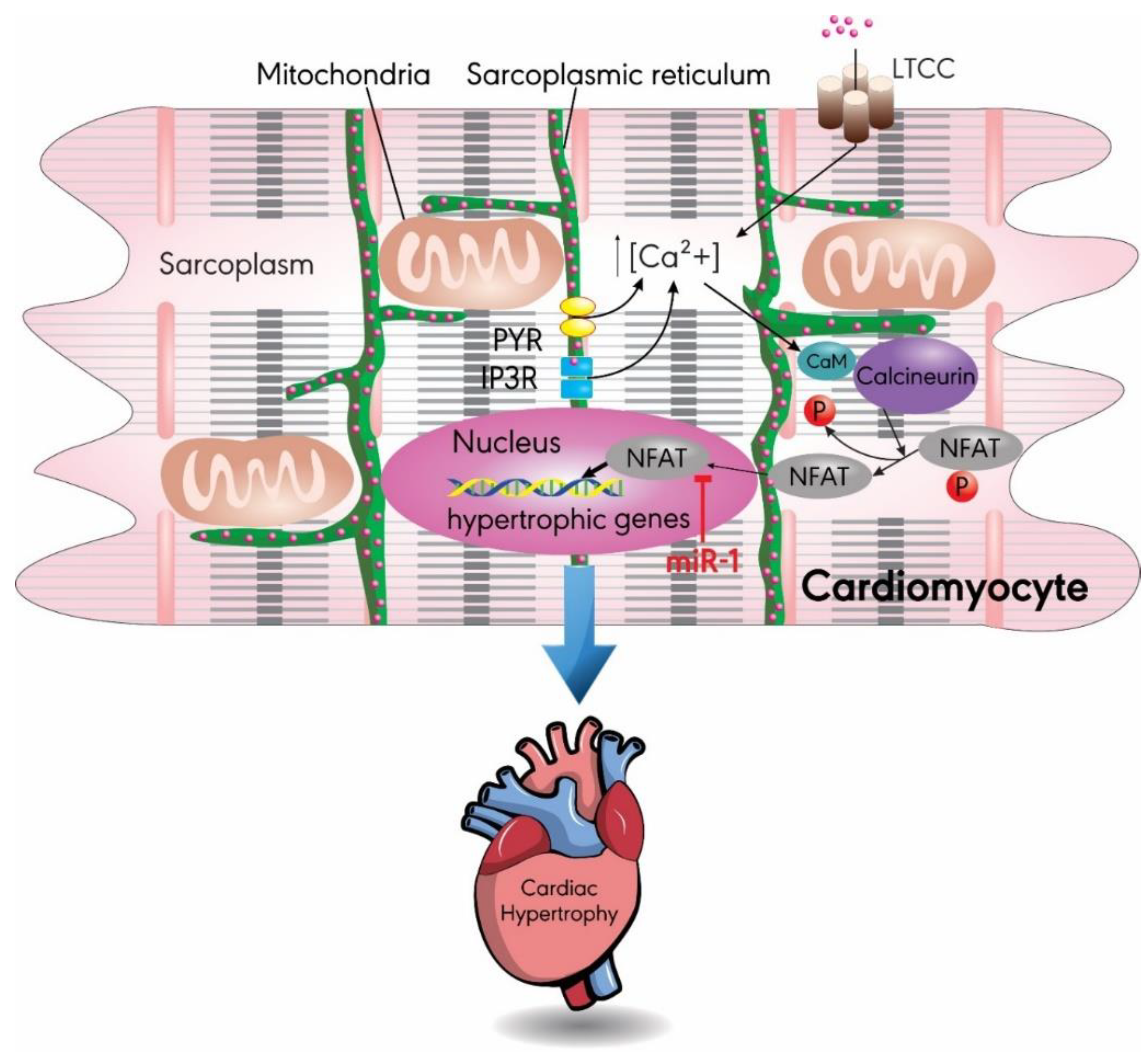
3.1. MiR-1
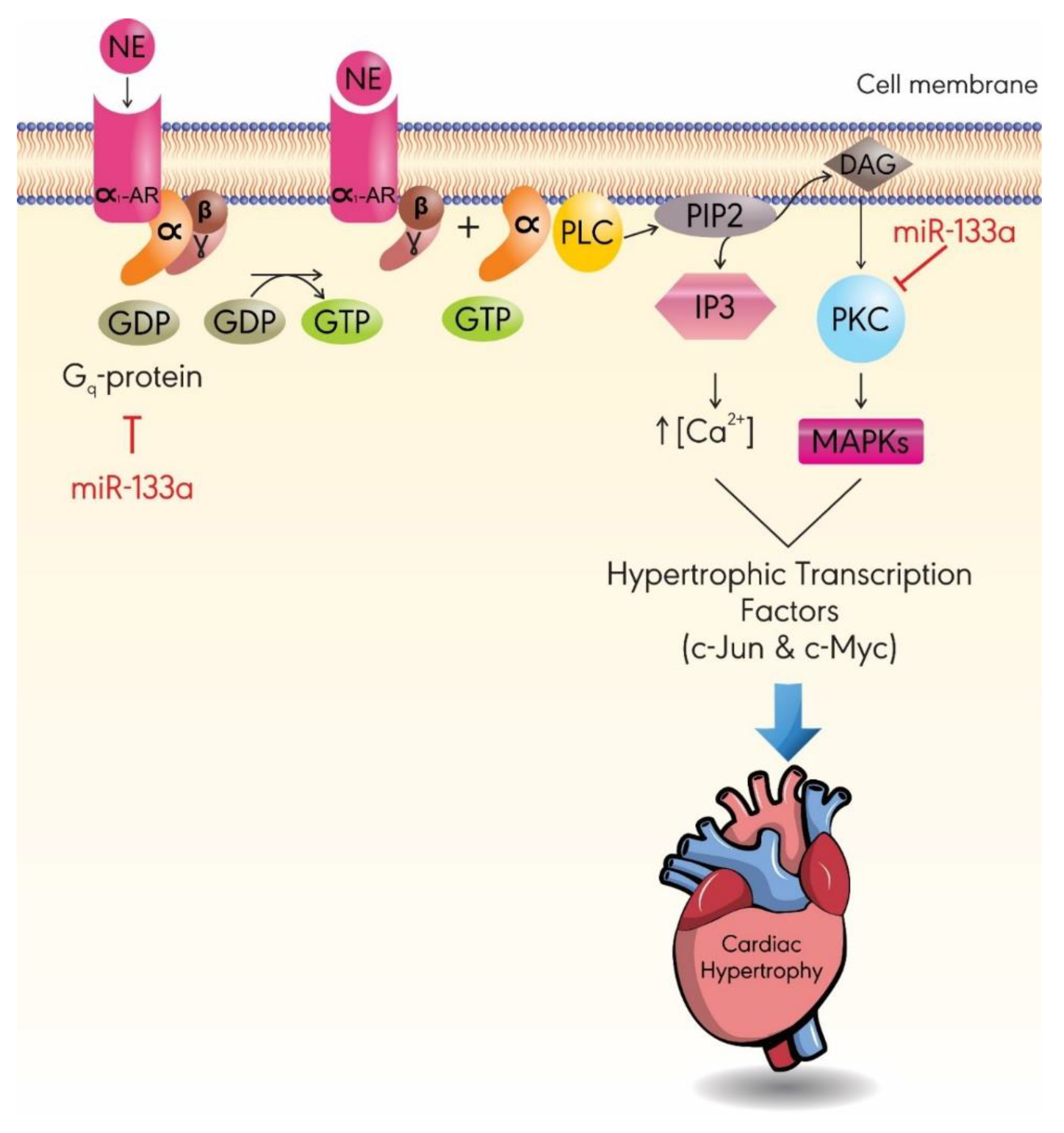
3.2. MiR-133a
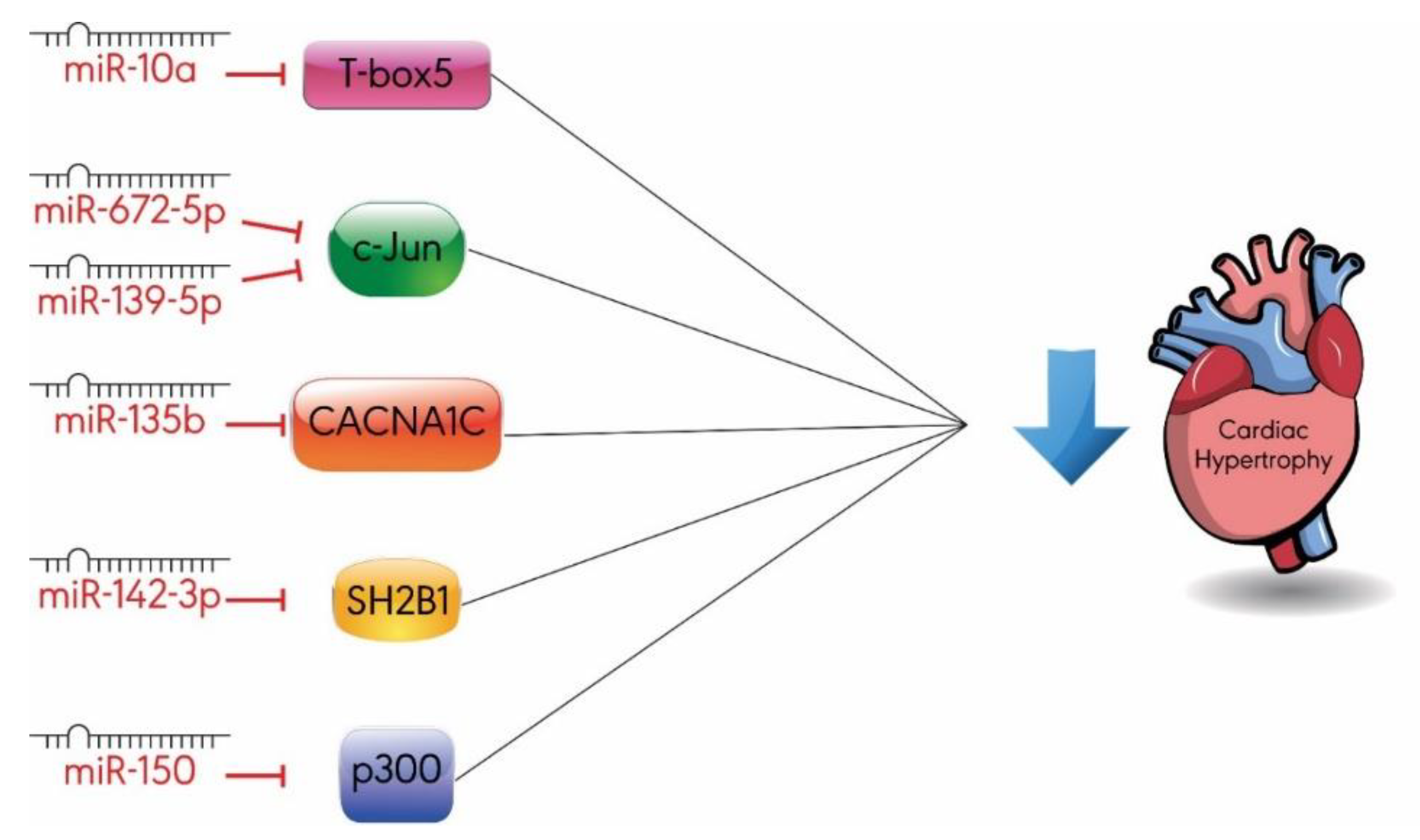
3.3. Others
4. MicroRNAs that Promote Cardiac Hypertrophy
4.1. MiR-155
4.2. MiR-22
4.3. MiR-217
4.4. MiR-29
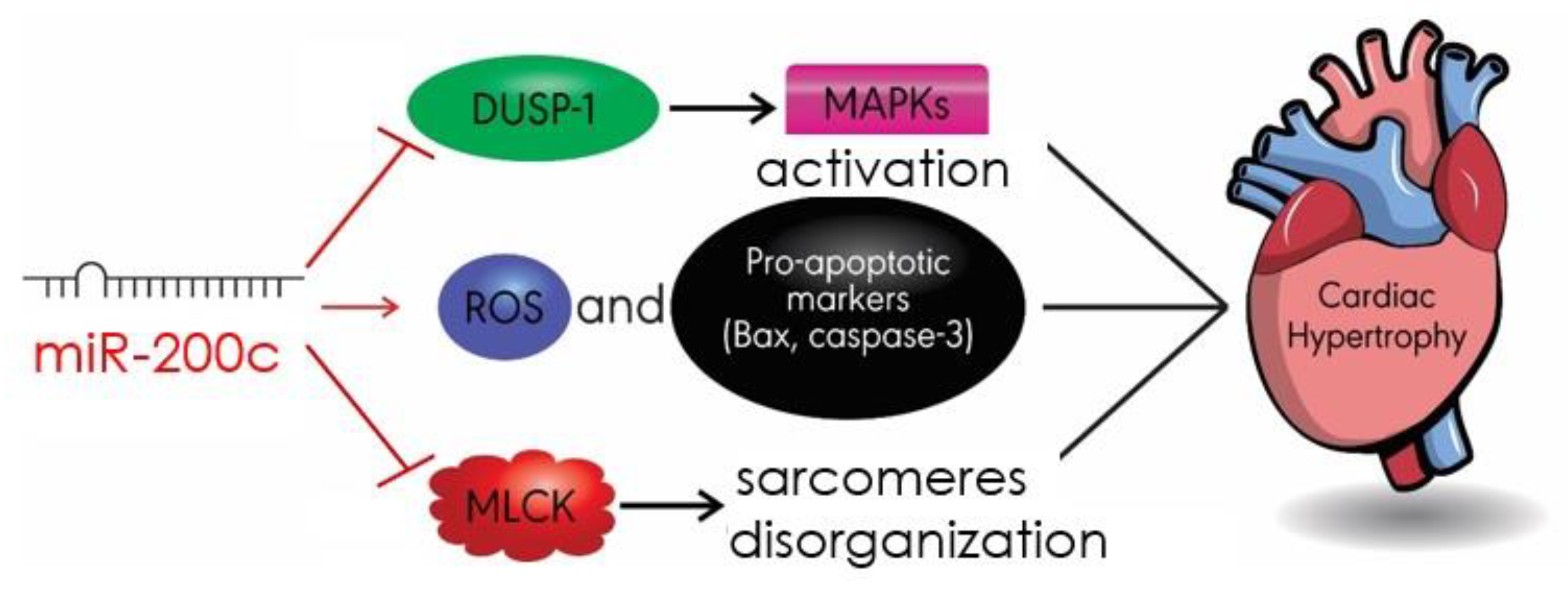
4.5. MiR-200c
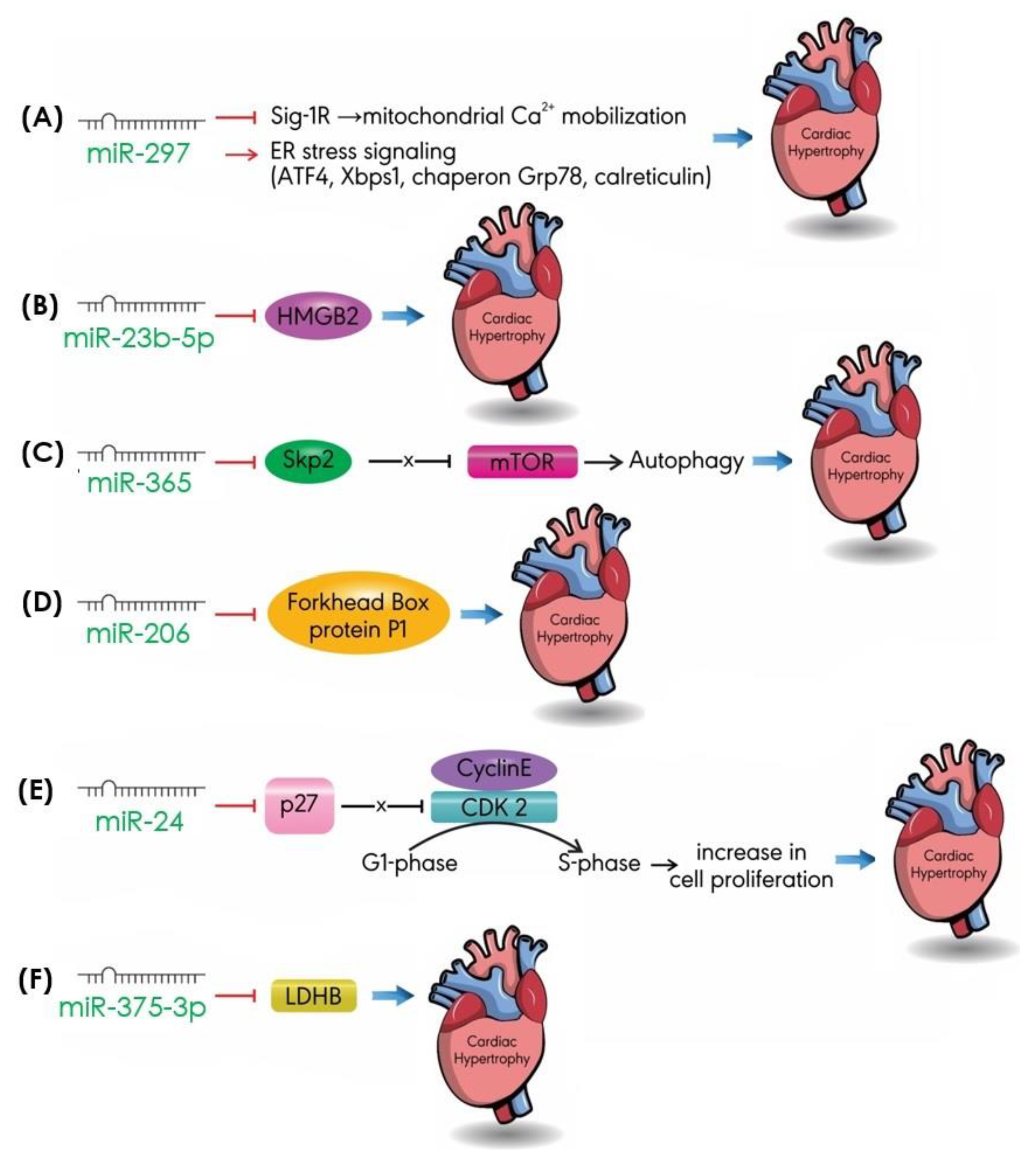
4.6. Others
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling-concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Lee, L.C.; Zhihong, Z.; Hinson, A.; Guccione, J.M. Reduction in left ventricular wall stress and improvement in function in failing hearts using Algisyl-LVR. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 2007, 49, 962–970. [Google Scholar] [CrossRef]
- Hirt, M.N.; Sorensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Vollert, I.; Stohr, A.; Schulze, T.; Witten, A.; et al. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Mihl, C.; Dassen, W.; Kuipers, H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth. Heart J. 2008, 16, 129–133. [Google Scholar] [CrossRef]
- Carabello, B.A. Concentric versus eccentric remodeling. J. Card. Fail. 2002, 8, S258–S263. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Bisping, E.; Wakula, P.; Poteser, M.; Heinzel, F.R. Targeting cardiac hypertrophy: Toward a causal heart failure therapy. J. Cardiovasc. Pharmacol. 2014, 64, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; An, X.; Niu, L. Role of microRNAs in cardiac development and disease. Exp. Ther. Med. 2017, 13, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Tatsuguchi, M.; Seok, H.Y.; Callis, T.E.; Thomson, J.M.; Chen, J.F.; Newman, M.; Rojas, M.; Hammond, S.M.; Wang, D.Z. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 1137–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, J.J.; Coller, H.A. The code within the code: microRNAs target coding regions. Cell Cycle 2010, 9, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, É.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, T.Y.; Cao, J.N.; Feng, Q.T.; Fu, Y.J.; Xu, X.; Yang, C.J. MicroRNA-206 Downregulates connexin43 in cardiomyocytes to induce cardiac arrhythmias in a transgenic mouse model. Heart Lung Circ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis 2018, 271, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derda, A.A.; Woo, C.C.; Wongsurawat, T.; Richards, M.; Lee, C.N.; Kofidis, T.; Kuznetsov, V.A.; Sorokin, V.A. Gene expression profile analysis of aortic vascular smooth muscle cells reveals upregulation of cadherin genes in myocardial infarction patients. Physiol. Genom. 2018, 50, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients. Data Brief 2018, 17, 1112–1135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Wang, J.; Wee, A.S.; Yong, Q.W.; Tay, E.L.; Woo, C.C.; Sorokin, V.; Richards, A.M.; Ling, L.H. Differential microRNA expression profile in myxomatous mitral valve prolapse and fibroelastic deficiency valves. Int. J. Mol. Sci. 2016, 17, 753. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovasc. Res. 2019, 115, 739–751. [Google Scholar] [CrossRef]
- Raso, A.; Dirkx, E.; Philippen, L.E.; Fernandez-Celis, A.; De Majo, F.; Sampaio-Pinto, V.; Sansonetti, M.; Juni, R.; El Azzouzi, H.; Calore, M.; et al. Therapeutic delivery of miR-148a suppresses ventricular dilation in HEart failure. Mol. Ther. 2019, 27, 584–599. [Google Scholar] [CrossRef]
- Boon, R.A.; Dimmeler, S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef]
- Creemers, E.E.; van Rooij, E. Function and therapeutic potential of noncoding RNAs in cardiac fibrosis. Circ. Res. 2016, 118, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y.T. Overview of microRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int. J. Mol. Sci. 2016, 17, 749. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kaminska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, Y.; Zhu, Y.; Zhang, Y.; Bei, Y. Noncoding RNAs in Cardiac Hypertrophy. J. Cardiovasc. Transl. Res. 2018, 11, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Xu, X.; Chen, Y.-H. The role of miR-1 in the heart: From cardiac morphogenesis to physiological function. Hum. Genet. Embryol. 2014, 4, 119. [Google Scholar]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Wilkins, B.J.; De Windt, L.J.; Bueno, O.F.; Braz, J.C.; Glascock, B.J.; Kimball, T.F.; Molkentin, J.D. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol. Cell. Biol. 2002, 22, 7603–7613. [Google Scholar] [CrossRef]
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, L.; Zhang, S.; Zhang, Y.; Lei, S. MicroRNA-1 suppresses cardiac hypertrophy by targeting nuclear factor of activated T cells cytoplasmic 3. Mol. Med. Rep. 2015, 12, 8282–8288. [Google Scholar] [CrossRef] [PubMed]
- Krenz, M.; Robbins, J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 2004, 44, 2390–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stolen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.; Tang, C.; Zhu, W.; Zhu, J.; Lin, Q.; Fu, Y.; Deng, C.; Xue, Y.; Yang, M.; Wu, S.; et al. CDK6 mediates the effect of attenuation of miR-1 on provoking cardiomyocyte hypertrophy. Mol. Cell. Biochem. 2016, 412, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Diniz, G.P.; Lino, C.A.; Moreno, C.R.; Senger, N.; Barreto-Chaves, M.L.M. MicroRNA-1 overexpression blunts cardiomyocyte hypertrophy elicited by thyroid hormone. J. Cell. Physiol. 2017, 232, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, R.; Hansen, A.H.; Haunso, S.; Busk, P.K. Phosphorylation of pRb by cyclin D kinase is necessary for development of cardiac hypertrophy. Cell Prolif. 2008, 41, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Song, X.W.; Zou, J.; Wang, G.K.; Kremneva, E.; Li, X.Q.; Zhu, N.; Sun, T.; Lappalainen, P.; Yuan, W.J.; et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J. Cell Sci. 2010, 123, 2444–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevalainen, E.M.; Skwarek-Maruszewska, A.; Braun, A.; Moser, M.; Lappalainen, P. Two biochemically distinct and tissue-specific twinfilin isoforms are generated from the mouse Twf2 gene by alternative promoter usage. Biochem. J. 2009, 417, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef]
- Latronico, M.V.; Costinean, S.; Lavitrano, M.L.; Peschle, C.; Condorelli, G. Regulation of cell size and contractile function by AKT in cardiomyocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 250–260. [Google Scholar] [CrossRef]
- Condorelli, G.; Drusco, A.; Stassi, G.; Bellacosa, A.; Roncarati, R.; Iaccarino, G.; Russo, M.A.; Gu, Y.; Dalton, N.; Chung, C.; et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 12333–12338. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.L.; Chen, C.; Huo, R.; Wang, N.; Li, Z.; Tu, Y.J.; Hu, J.T.; Chu, X.; Huang, W.; Yang, B.F. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: A novel mechanism for progressive cardiac hypertrophy. Hypertension 2010, 55, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Liu, L.H.; Oiseth, S.K.; Beloy, J.; Lundin, L.; Gidley, M.J.; Day, L. Influence of boron on carrot cell wall structure and its resistance to fracture. J. Agric. Food Chem. 2010, 58, 9181–9189. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Lee, C.Y.; Ham, O.; Moon, J.Y.; Lee, J.; Seo, H.-H.; Shin, S.; Kim, S.W.; Lee, S.; Lim, S. MicroRNA-133a attenuates cardiomyocyte hypertrophy by targeting PKCδ and Gq. Mol. Cell. Biochem. 2018, 439, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, S.; George, B.; Feng, Q.; Chakrabarti, S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab. Res. Rev. 2010, 26, 40–49. [Google Scholar] [CrossRef]
- Diniz, G.P.; Lino, C.A.; Guedes, E.C.; do Nascimento Moreira, L.; Barreto-Chaves, M.L.M. Cardiac microRNA-133 is down-regulated in thyroid hormone-mediated cardiac hypertrophy partially via Type 1 angiotensin II receptor. Basic Res. Cardiol. 2015, 110, 49. [Google Scholar] [CrossRef]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 126–137. [Google Scholar] [CrossRef]
- Filtz, T.M.; Grubb, D.R.; McLeod-Dryden, T.J.; Luo, J.; Woodcock, E.A. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cbeta1b. FASEB 2009, 23, 3564–3570. [Google Scholar] [CrossRef]
- Alcantara-Hernandez, R.; Hernandez-Mendez, A. Adrenergic signaling molecular complexes. Gac. Med. Mex. 2018, 154, 223–235. [Google Scholar] [CrossRef]
- Wang, D.; Zhai, G.; Ji, Y.; Jing, H. MicroRNA-10a targets T-box 5 to inhibit the development of cardiac hypertrophy. Int. Heart. J. 2017, 58, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Li, A.; Chen, X.; Qin, Y.; Sun, X.; Li, Y.; Yue, E.; Wang, C.; Ding, X.; Yan, Y.; et al. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int. J. Cardiol. 2018, 269, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Cheng, M.; Hu, S.; Wang, S.; Wang, L.; Tu, X.; Huang, C.X.; Jiang, H.; Wu, G. Overexpression of miR-142-3p improves mitochondrial function in cardiac hypertrophy. Biomed. Pharm. 2018, 108, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; van Leeuwen, R.E.; Custers, K.; Peters, T.; Hazebroek, M.; Stoger, L.; Wijnands, E.; et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.Y.; Chen, J.; Kataoka, M.; Huang, Z.P.; Ding, J.; Yan, J.; Hu, X.; Wang, D.Z. Loss of MicroRNA-155 protects the heart from pathological cardiac hypertrophy. Circ. Res. 2014, 114, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y.; Cao, Z.; Tong, X.Z.; Xie, H.Q.; Luo, T.; Hua, X.P.; Wang, H.Q. miR-155 functions downstream of angiotensin II receptor subtype 1 and calcineurin to regulate cardiac hypertrophy. Exp. Ther. Med. 2016, 12, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wu, F. A new miRNA regulator, miR-672, reduces cardiac hypertrophy by inhibiting JUN expression. Gene 2018, 648, 21–30. [Google Scholar] [CrossRef]
- Ming, S.; Shui-Yun, W.; Wei, Q.; Jian-Hui, L.; Ru-Tai, H.; Lei, S.; Mei, J.; Hui, W.; Ji-Zheng, W. MiR-139-5p inhibits isoproterenol-induced cardiac hypertrophy by targetting c-Jun. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Duan, Y.; Zhou, B.; Su, H.; Liu, Y.; Du, C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp. Cell Res. 2013, 319, 173–184. [Google Scholar] [CrossRef]
- Kim, T.G.; Chen, J.; Sadoshima, J.; Lee, Y. Jumonji represses atrial natriuretic factor gene expression by inhibiting transcriptional activities of cardiac transcription factors. Mol. Cell. Biol. 2004, 24, 10151–10160. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Shirato, H.; Nakajima, K.; Kojima, M.; Takahashi, M.; Kubota, M.; Suzuki-Migishima, R.; Motegi, Y.; Yokoyama, M.; Takeuchi, T. jumonji downregulates cardiac cell proliferation by repressing cyclin D1 expression. Dev. Cell 2003, 5, 85–97. [Google Scholar] [CrossRef]
- Lunde, I.G.; Kvaloy, H.; Austbo, B.; Christensen, G.; Carlson, C.R. Angiotensin II and norepinephrine activate specific calcineurin-dependent NFAT transcription factor isoforms in cardiomyocytes. J. Appl. Physiol. 2011, 111, 1278–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.P.; Chen, J.; Seok, H.Y.; Zhang, Z.; Kataoka, M.; Hu, X.; Wang, D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013, 112, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Kook, H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar] [CrossRef]
- Xu, X.D.; Song, X.W.; Li, Q.; Wang, G.K.; Jing, Q.; Qin, Y.W. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J. Cell. Physiol. 2012, 227, 1391–1398. [Google Scholar] [CrossRef]
- Tu, Y.; Wan, L.; Bu, L.; Zhao, D.; Dong, D.; Huang, T.; Cheng, Z.; Shen, B. MicroRNA-22 downregulation by atorvastatin in a mouse model of cardiac hypertrophy: A new mechanism for antihypertrophic intervention. Cell. Physiol. Biochem. 2013, 31, 997–1008. [Google Scholar] [CrossRef]
- Nie, X.; Fan, J.; Li, H.; Yin, Z.; Zhao, Y.; Dai, B.; Dong, N.; Chen, C.; Wang, D.W. MiR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol. Ther. Nucleic acids 2018, 12, 254–266. [Google Scholar] [CrossRef]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2017, 127, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, M.; Nakajima, K.; Makino, T.; Ogawa, S.; Kojima, M.; Ito, S.; Ikenishi, A.; Hayashi, T.; Schwartz, R.J.; Nakamura, K.; et al. Histone H3 lysine 9 methyltransferases, G9a and GLP are essential for cardiac morphogenesis. Mech. Dev. 2013, 130, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Avramopoulos, P.; Ramanujam, D.; Gruter, L.; Werfel, S.; Giosele, S.; Brunner, A.D.; Esfandyari, D.; Papadopoulou, A.S.; De Strooper, B.; et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun. 2017, 8, 1614. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Y.; Chen, C.; Xu, X.; Lu, Q. miR-29a promotes pathological cardiac hypertrophy by targeting the PTEN/AKT/mTOR signalling pathway and suppressing autophagy. Acta Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, Z.; Dai, F.F.; Wang, H.; Zhou, M.J.; Yang, M.H.; Zhang, S.F.; Fu, Z.F.; Mei, Y.W.; Zang, M.X.; et al. MiR-29a attenuates cardiac hypertrophy through inhibition of PPARdelta expression. J. Cell. Physiol. 2019, 234, 13252–13262. [Google Scholar] [CrossRef]
- Li, M.; Wang, N.; Zhang, J.; He, H.P.; Gong, H.Q.; Zhang, R.; Song, T.F.; Zhang, L.N.; Guo, Z.X.; Cao, D.S.; et al. MicroRNA-29a-3p attenuates ET-1-induced hypertrophic responses in H9c2 cardiomyocytes. Gene 2016, 585, 44–50. [Google Scholar] [CrossRef]
- Singh, G.B.; Raut, S.K.; Khanna, S.; Kumar, A.; Sharma, S.; Prasad, R.; Khullar, M. MicroRNA-200c modulates DUSP-1 expression in diabetes-induced cardiac hypertrophy. Mol. Cell. Biochem. 2017, 424, 1–11. [Google Scholar] [CrossRef]
- Hu, S.; Cheng, M.; Guo, X.; Wang, S.; Liu, B.; Jiang, H.; Huang, C.; Wu, G. Down-regulation of miR-200c attenuates AngII-induced cardiac hypertrophy via targeting the MLCK-mediated pathway. J. Cell. Mol. Med. 2019, 23, 2505–2516. [Google Scholar] [CrossRef]
- Bueno, O.F.; De Windt, L.J.; Lim, H.W.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Molkentin, J.D. The dual-specificity phosphatase MKP-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ. Res. 2001, 88, 88–96. [Google Scholar] [CrossRef]
- Chan, J.Y.; Takeda, M.; Briggs, L.E.; Graham, M.L.; Lu, J.T.; Horikoshi, N.; Weinberg, E.O.; Aoki, H.; Sato, N.; Chien, K.R.; et al. Identification of cardiac-specific myosin light chain kinase. Circ. Res. 2008, 102, 571–580. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, S.; Guo, C.; Li, J.; Sang, W. Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. Int. J. Mol. Med. 2017, 39, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Chen, L.; Li, J.; Zhao, M.; Wu, S.; Wu, W.; Liu, X. Role of microRNA-124 in cardiomyocyte hypertrophy induced by angiotensin II. Cell. Mol. Biol. 2017, 63, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Wang, X.; Li, R.; Yin, D. MicroRNA-365 accelerates cardiac hypertrophy by inhibiting autophagy via the modulation of Skp2 expression. Biochem. Biophys. Res. Commun. 2017, 484, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wu, J.; Chen, P.; Wang, J.; Deng, Y.; Zhu, G.; Xian, J.; Huang, L.; Ouyang, W. MicroRNA-375-3p inhibitor suppresses angiotensin II-induced cardiomyocyte hypertrophy by promoting lactate dehydrogenase B expression. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Shinoda, Y.; Tagashira, H.; Bhuiyan, M.S.; Hasegawa, H.; Kanai, H.; Fukunaga, K. Haloperidol aggravates transverse aortic constriction-induced heart failure via mitochondrial dysfunction. J. Pharmacol. Sci. 2016, 131, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Bao, Q.; Zhao, M.; Chen, L.; Wang, Y.; Wu, S.; Wu, W.; Liu, X. MicroRNA-297 promotes cardiomyocyte hypertrophy via targeting sigma-1 receptor. Life Sci. 2017, 175, 1–10. [Google Scholar] [CrossRef]
- Boureima Oumarou, D.; Ji, H.; Xu, J.; Li, S.; Ruan, W.; Xiao, F.; Yu, F. Involvement of microRNA-23b-5p in the promotion of cardiac hypertrophy and dysfunction via the HMGB2 signaling pathway. Biomed. Pharm. 2019, 116, 108977. [Google Scholar] [CrossRef]
- Wang, L.L.; Meng, Q.H.; Jiao, Y.; Xu, J.Y.; Ge, C.M.; Zhou, J.Y.; Rosen, E.M.; Wang, H.C.; Fan, S.J. High-mobility group boxes mediate cell proliferation and radiosensitivity via retinoblastoma-interaction-dependent and -independent mechanisms. Cancer Biother. Radiopharm. 2012, 27, 329–335. [Google Scholar] [CrossRef]
- Jentzsch, C.; Leierseder, S.; Loyer, X.; Flohrschutz, I.; Sassi, Y.; Hartmann, D.; Thum, T.; Laggerbauer, B.; Engelhardt, S. A phenotypic screen to identify hypertrophy-modulating microRNAs in primary cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 13–20. [Google Scholar] [CrossRef]
- Yang, Y.; Del Re, D.P.; Nakano, N.; Sciarretta, S.; Zhai, P.; Park, J.; Sayed, D.; Shirakabe, A.; Matsushima, S.; Park, Y.; et al. MiR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ. Res. 2015, 117, 891–904. [Google Scholar] [CrossRef]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, M.; Liu, R.F.; Zhang, J.S.; Xu, M. Cardiac hypertrophy is positively regulated by microRNA24 in rats. Chin. Med. J. 2018, 131, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Dehaini, H.; Awada, H.; El-Yazbi, A.; Zouein, F.A.; Issa, K.; Eid, A.A.; Ibrahim, M.; Badran, A.; Baydoun, E.; Pintus, G.; et al. MicroRNAs as potential pharmaco-targets in ischemia-reperfusion injury compounded by diabetes. Cells 2019, 8, 152. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MiRNAs | Targets | Signaling Pathway | References |
|---|---|---|---|
| Anti-hypertrophic | |||
| miR-1 | Calcineurin MCU CDK6-Rb HDAC4 TWF1 IGF-1 | Calcium signaling Calcium signaling CDK-Rb pathway Transcription Actin monomer size and localization Transcription | [41] [43] [44] [45] [47] [49] |
| miR-133a | Calcineurin NFATC4 PLC-PKC SERCA2a SRF/cyclin D2 IGF-1R/SGK1/MEF2 | Calcium signaling Calcium signaling Calcium signaling/Transcription Calcium signaling Cell cycle MAPK/ERK | [52] [53] [57] [54] [55] [56] |
| miR-10a | T-box5 | Transcription | [61] |
| miR-672-5p | c-Jun | Transcription | [68] |
| miR-139-5p | c-Jun | Transcription | [69] |
| miR-135b | L-type Ca2+ channels | Calcium signaling | [62] |
| miR-142-3p | SH2B1 | Energy balance | [63] |
| miR-150 | p300 | Transcription | [70] |
| Pro-hypertrophic | |||
| miR-155 | SOCS1 Jarid2 AT1R | JAK/STAT3 Calcineurin Calcium signaling | [64] [65] [73] |
| miR-22 | HDAC4/SIRT1 PTEN | Calcineurin PI3K/Akt/mTOR | [74] [78,79] |
| miR-217 | PTEN H3K9me2/EHMT1 &2 | PI3K/Akt/mTOR Transcription | [80] [81] |
| miR-29 | GSK3B, ICAT/CTNNBIP1, HBP1, GLIS2 PTEN | Wnt signaling PI3K/Akt/mTOR | [83] [84] |
| miR-200c | DUSP-1 MLCK Bax/cleaved caspase3 | MAPK/JANK/p38 Apoptosis | [87] [88] [88] |
| miR-297 | Sig-1R ATF4, Xbps1, Chaperon G78, Calreticulin | Mitochondrial Ca2+ mobilization ER stress signaling pathway | [96] [96] |
| miR-124 | ER stress markers | ER stress signaling pathway | [92] |
| miR- 23b-5p | HMGB2 | Transcription/autophagy | [97] |
| miR-365 | Skp2 | mTOR | [93] |
| miR-206 | Forkhead box protein P1 | Transcription | [100] |
| miR-24 | p27 | Cell cycle | [104] |
| miR-375-3p | LDHB | Cell metabolism | [94] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wehbe, N.; Nasser, S.A.; Pintus, G.; Badran, A.; Eid, A.H.; Baydoun, E. MicroRNAs in Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 4714. https://doi.org/10.3390/ijms20194714
Wehbe N, Nasser SA, Pintus G, Badran A, Eid AH, Baydoun E. MicroRNAs in Cardiac Hypertrophy. International Journal of Molecular Sciences. 2019; 20(19):4714. https://doi.org/10.3390/ijms20194714
Chicago/Turabian StyleWehbe, Nadine, Suzanne A. Nasser, Gianfranco Pintus, Adnan Badran, Ali H. Eid, and Elias Baydoun. 2019. "MicroRNAs in Cardiac Hypertrophy" International Journal of Molecular Sciences 20, no. 19: 4714. https://doi.org/10.3390/ijms20194714
APA StyleWehbe, N., Nasser, S. A., Pintus, G., Badran, A., Eid, A. H., & Baydoun, E. (2019). MicroRNAs in Cardiac Hypertrophy. International Journal of Molecular Sciences, 20(19), 4714. https://doi.org/10.3390/ijms20194714