Progress in iPSC-Based Modeling of Psychiatric Disorders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
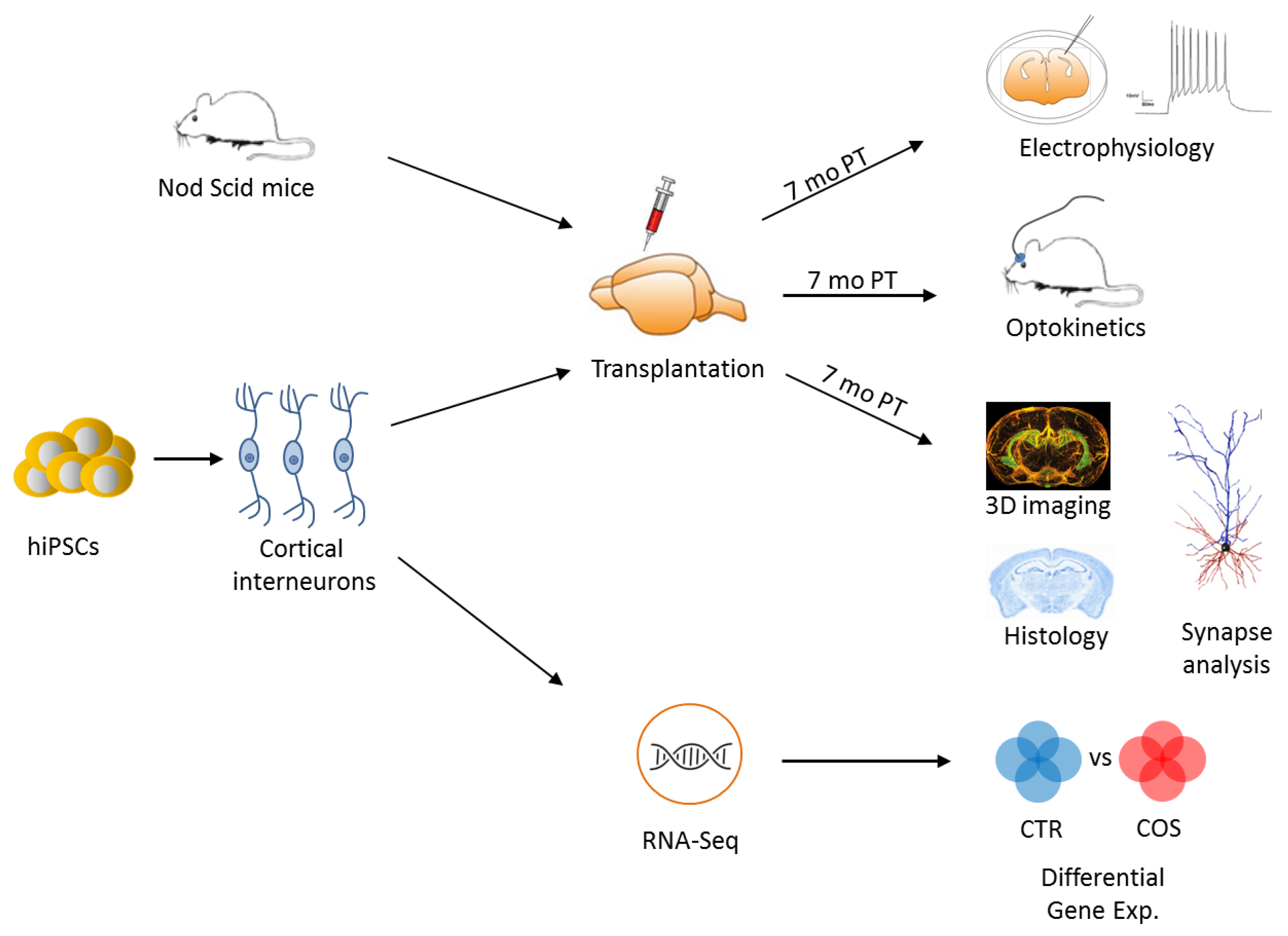
2. Reconstructing Human Brain Development and Circuitries In Vitro
3. In Vivo Studies of Patient-Specific iPSCs
4. Discussion and Outlook
4.1. Beyond 2D-Cell Culture
4.2. Building Neural Circuits In Vitro
4.3. Transplantation of iPSC Derived Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BD | Bipolar disorder |
| CA3 | cornu ammonis subfield 3 |
| COS | childhood onset of schizophrenia |
| ESC | embryonic stem cell |
| DG | dentate gyrus |
| GABA | γ-aminobutyric |
| cIN | cortical interneuron |
| hGPC | human glial progenitor cells |
| hiMG | human induced microglia |
| iPSC | induced pluripotent stem cells |
| NPC | neural progenitor cells |
| MD | major depression |
| SCZ | schizophrenia |
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet (London, England) 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Olfson, M.; Gerhard, T.; Huang, C.; Crystal, S.; Stroup, T.; Olfson, M.M.; Gerhard, P.T.; Huang, P.C.; Crystal, P.S. Premature Mortality Among Adults With Schizophrenia in the United States. JAMA Psychiatry 2015, 72, 1. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, B. Schizophrenia (3rd Edition) Daniel R. Weinberger (Editor), Paul Harrison (Editor) Chichester: Wiley-Blackwell, 2011. ISBN: 978-1-4051-7697-2. Ir. J. Psychol. Med. 2014, 29, 200. [Google Scholar] [CrossRef]
- Cannon, T.D.; Thompson, P.M.; Van Erp, T.G.M.; Toga, A.W.; Poutanen, V.-P.; Huttunen, M.; Lonnqvist, J.; Standerskjold-Nordenstam, C.-G.; Narr, K.L.; Khaledy, M.; et al. Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 3228–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhshi, K.; Chance, S.A. The neuropathology of schizophrenia: A selective review of past studies and emerging themes in brain structure and cytoarchitecture. Neuroscience 2015, 303, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J. Postmortem studies in schizophrenia. Dialogues Clin. Neurosci. 2000, 2, 349–357. [Google Scholar] [PubMed]
- Strakowski, S.M. The Bipolar Brain: Integrating Neuroimaging with Genetics; Oxford University Press: New York, NY, USA, 2012; ISBN 978-0-19-979760-8. [Google Scholar]
- Hyman, S.E. Back to basics: Luring industry back into neuroscience. Nat. Neurosci. 2016, 19, 1383–1384. [Google Scholar] [CrossRef] [PubMed]
- Vahia, V.N. Diagnostic and statistical manual of mental disorders 5: A quick glance. Indian J. Psychiatry 2013, 55, 220–223. [Google Scholar] [CrossRef]
- Stephan, K.E.; Bach, D.R.; Fletcher, P.C.; Flint, J.; Frank, M.J.; Friston, K.J.; Heinz, A.; Huys, Q.J.M.; Owen, M.J.; Binder, E.B.; et al. Charting the landscape of priority problems in psychiatry, part 1: Classification and diagnosis. Lancet Psychiatry 2016, 3, 77–83. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef]
- McIntosh, A.M.; Sullivan, P.F.; Lewis, C.M. Uncovering the Genetic Architecture of Major Depression. Neuron 2019, 102, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Geschwind, D.H. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019, 177, 162–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.J.; O’Donovan, M.C. Schizophrenia and the neurodevelopmental continuum:evidence from genomics. World Psychiatry 2017, 16, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, R.; Weinberger, D.R. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 2017, 18, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, I.I.; Gould, T.D. The Endophenotype Concept in Psychiatry: Etymology and Strategic Intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Ahmad, R.; Sportelli, V.; Ziller, M.; Spengler, D.; Hoffmann, A. Tracing Early Neurodevelopment in Schizophrenia with Induced Pluripotent Stem Cells. Cells 2018, 7, 140. [Google Scholar] [CrossRef]
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish. Int. J. Mol. Sci. 2018, 19, 770. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ziller, M.; Spengler, D. Childhood-Onset Schizophrenia: Insights from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 3829. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G.; Arlotta, P. Present and future of modeling human brain development in 3D organoids. Curr. Opin. Cell Biol. 2017, 49, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, E.K.; Benson, C.A.; Narla, S.T.; Dimitri, A.; Chuye, L.E.B.; Dhiman, S.; Harikrishnan, K.; Elahi, S.; Freedman, D.; Brennand, K.J.; et al. Cerebral organoids reveal early cortical maldevelopment in schizophrenia-computational anatomy and genomics, role of FGFR1. Transl. Psychiatry 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.X.; Di Giorgio, F.P.; Yao, J.; Marchetto, M.C.; Brennand, K.; Wright, R.; Mei, A.; McHenry, L.; Lisuk, D.; Grasmick, J.M.; et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2014, 2, 295–310. [Google Scholar] [CrossRef]
- Wadehra, S.; Pruitt, P.; Murphy, E.R.; Diwadkar, V.A. Network dysfunction during associative learning in schizophrenia: Increased activation, but decreased connectivity: An fMRI study. Schizophr. Res. 2013, 148, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Mei, A.; Paquola, A.C.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S.; Neshat, N.; Kim, H.J.; Ku, M.; et al. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697.e9. [Google Scholar] [Green Version]
- Hook, V.; Brennand, K.J.; Kim, Y.; Toneff, T.; Funkelstein, L.; Lee, K.C.; Ziegler, M.; Gage, F.H. Human iPSC neurons display activity-dependent neurotransmitter secretion: Aberrant catecholamine levels in schizophrenia neurons. Stem Cell Rep. 2014, 3, 531–538. [Google Scholar] [CrossRef]
- Raabe, F.J.; Galinski, S.; Papiol, S.; Falkai, P.G.; Schmitt, A.; Rossner, M.J. Studying and modulating schizophrenia-associated dysfunctions of oligodendrocytes with patient-specific cell systems. npj Schizophr. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, M.; Magistretti, P.J. A New Outlook on Mental Illnesses: Glial Involvement Beyond the Glue. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef]
- Forsyth, J.K.; Lewis, D.A. Mapping the Consequences of Impaired Synaptic Plasticity in Schizophrenia through Development: An Integrative Model for Diverse Clinical Features. Trends Cogn. Sci. 2017, 21, 760–778. [Google Scholar] [CrossRef]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sellgren, C.M.; Sheridan, S.D.; Gracias, J.; Xuan, D.; Fu, T.; Perlis, R.H. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 2017, 22, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, L.; Gage, F.H. Microglia, complement and schizophrenia. Nat. Neurosci. 2019, 22, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Bryois, J.; Skene, N.G.; Folkmann Hansen, T.; Kogelman, L.J.A.; Watson, H.J.; Brueggeman, L.; Breen, G.; Bulik, C.M.; Arenas, E.; Hjerling-Leffler, J.; et al. Genetic Identification of Cell Types Underlying Brain Complex Traits Yields Novel Insights Into the Etiology of Parkinson’s Disease. bioRxiv 2019, 528463. [Google Scholar] [CrossRef]
- Skene, N.G.; Bryois, J.; Bakken, T.E.; Breen, G.; Crowley, J.J.; Gaspar, H.A.; Giusti-Rodriguez, P.; Hodge, R.D.; Miller, J.A.; Muñoz-Manchado, A.B.; et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018, 50, 825–833. [Google Scholar] [CrossRef]
- Collado-Torres, L.; Burke, E.E.; Peterson, A.; Shin, J.; Straub, R.E.; Rajpurohit, A.; Semick, S.A.; Ulrich, W.S.; Price, A.J.; Valencia, C.; et al. Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron 2019, 103, 203–216.e8. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Steiner, J.; Guest, P.C.; Dobrowolny, H.; Bogerts, B. Glial cells as key players in schizophrenia pathology: Recent insights and concepts of therapy. Schizophr. Res. 2015, 161, 4–18. [Google Scholar] [CrossRef]
- Cassoli, J.S.; Guest, P.C.; Malchow, B.; Schmitt, A.; Falkai, P.; Martins-De-Souza, D. Disturbed macro-connectivity in schizophrenia linked to oligodendrocyte dysfunction: From structural findings to molecules. NPJ Schizophr. 2015, 1, 15034. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Cornea, E.; Murphy, V.; Styner, M.; Jarskog, L.F.; Gilmore, J.H. White matter development in infants at risk for schizophrenia. Schizophr. Res. 2019, 210, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Gogtay, N. Cortical brain development in schizophrenia: Insights from neuroimaging studies in childhood-onset schizophrenia. Schizophr. Bull. 2008, 34, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Addington, A.M.; Rapoport, J.L. The genetics of childhood-onset schizophrenia: When madness strikes the prepubescent. Curr. Psychiatry Rep. 2009, 11, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208.e6. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Glia keep synapse distribution under wraps. Cell 2013, 154, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Chen, M.; Wang, F.; Windrem, M.; Wang, S.; Shanz, S.; Xu, Q.; Oberheim, N.A.; Bekar, L.; Betstadt, S.; et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 2013, 12, 342–353. [Google Scholar] [CrossRef]
- Liu, Z.; Osipovitch, M.; Benraiss, A.; Huynh, N.P.T.; Foti, R.; Bates, J.; Chandler-Militello, D.; Findling, R.L.; Tesar, P.J.; Nedergaard, M.; et al. Dysregulated Glial Differentiation in Schizophrenia May Be Relieved by Suppression of SMAD4- and REST-Dependent Signaling. Cell Rep. 2019, 27, 3832–3843.e6. [Google Scholar] [CrossRef] [Green Version]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Volk, D.W.; Lewis, D.A. Early Developmental Disturbances of Cortical Inhibitory Neurons: Contribution to Cognitive Deficits in Schizophrenia. Schizophr. Bull. 2014, 40, 952–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Noh, H.; Bin Kim, W.; Ni, P.; Nguyen, C.; Cote, S.E.; Noyes, E.; Zhao, J.; Parsons, T.; Park, J.M.; et al. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell–derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci. 2019, 22, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Yagi, T. Clustered protocadherins and neuronal diversity. Prog. Mol. Biol. Transl. Sci. 2013, 116, 145–167. [Google Scholar] [PubMed]
- Soldner, F.; Jaenisch, R. Stem Cells, Genome Editing, and the Path to Translational Medicine. Cell 2018, 175, 615–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Yang, S.M.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Paşca, S.P. Assembling human brain organoids. Science 2019, 363, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Schrode, N.; Flaherty, E.; Brennand, K.J. New considerations for hiPSC-based models of neuropsychiatric disorders. Mol. Psychiatry 2018, 24, 49–66. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Narla, S.T.; Lee, Y.-W.; Benson, C.; Sarder, P.; Brennand, K.; Stachowiak, E.; Stachowiak, M. Common developmental genome deprogramming in schizophrenia - Role of Integrative Nuclear FGFR1 Signaling (INFS). Schizophr. Res. 2017, 185, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Real, R.; Peter, M.; Trabalza, A.; Khan, S.; Smith, M.A.; Dopp, J.; Barnes, S.J.; Momoh, A.; Strano, A.; Volpi, E.; et al. In vivo modeling of human neuron dynamics and Down syndrome. Science 2018, 362, eaau1810. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Ziller, M.; Spengler, D. Progress in iPSC-Based Modeling of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 4896. https://doi.org/10.3390/ijms20194896
Hoffmann A, Ziller M, Spengler D. Progress in iPSC-Based Modeling of Psychiatric Disorders. International Journal of Molecular Sciences. 2019; 20(19):4896. https://doi.org/10.3390/ijms20194896
Chicago/Turabian StyleHoffmann, Anke, Michael Ziller, and Dietmar Spengler. 2019. "Progress in iPSC-Based Modeling of Psychiatric Disorders" International Journal of Molecular Sciences 20, no. 19: 4896. https://doi.org/10.3390/ijms20194896
APA StyleHoffmann, A., Ziller, M., & Spengler, D. (2019). Progress in iPSC-Based Modeling of Psychiatric Disorders. International Journal of Molecular Sciences, 20(19), 4896. https://doi.org/10.3390/ijms20194896