Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation

Abstract
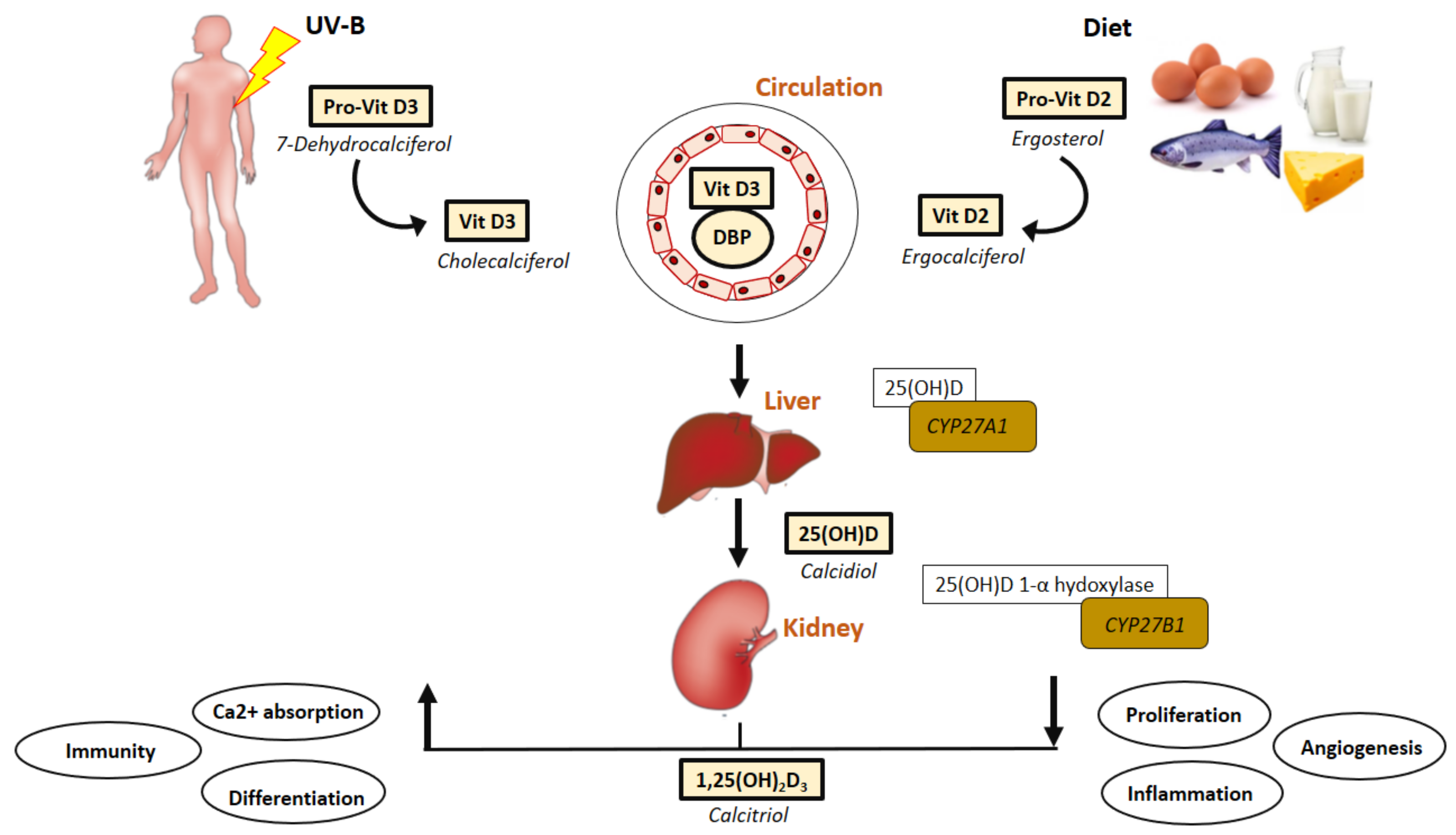
:1. Introduction
2. Peripheral Arterial Disease (PAD)
2.1. Vitamin D Status and PAOD Formation
2.2. Vitamin D Status and AAA Formation
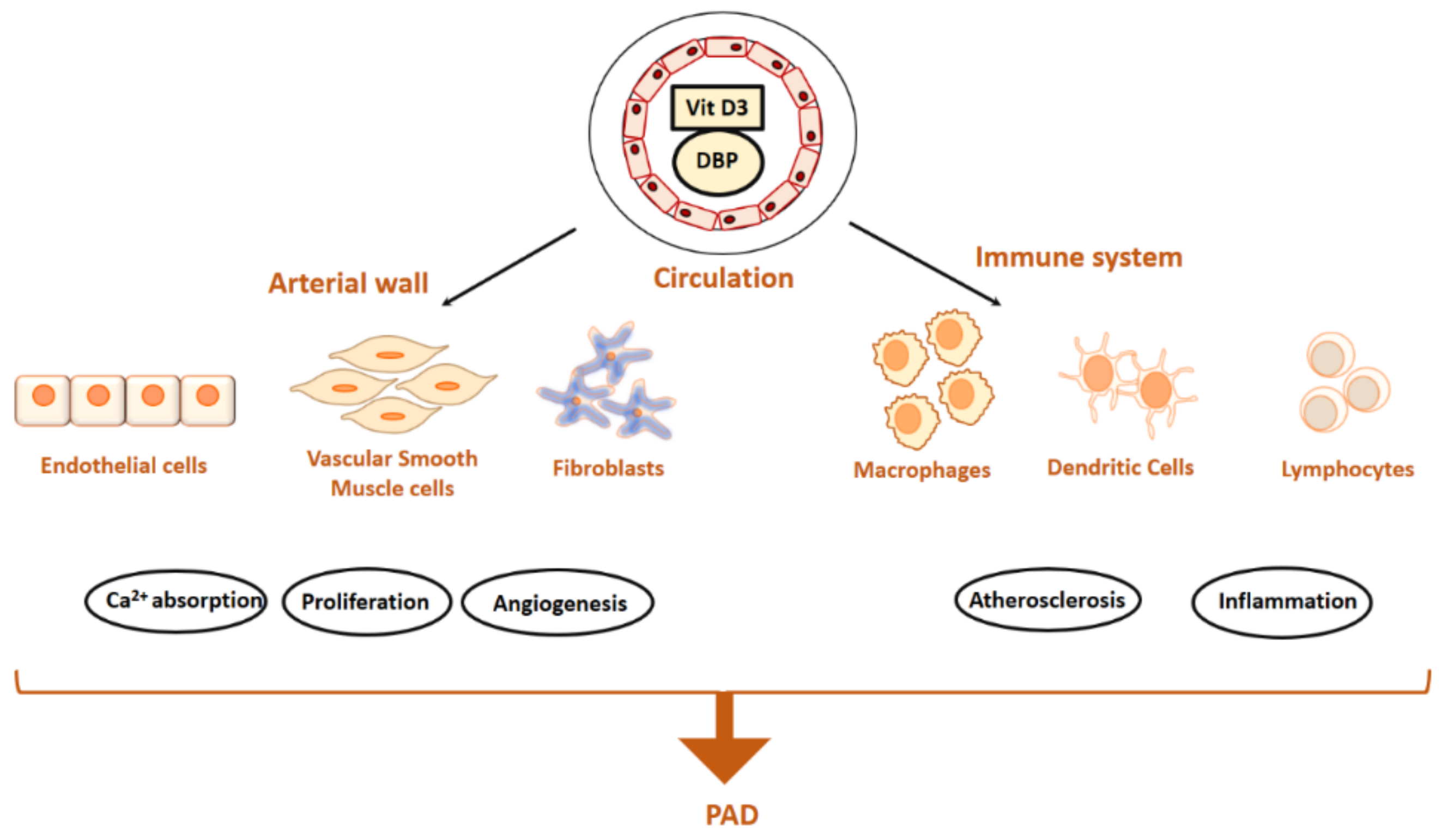
3. Vitamin D Status and Mechanisms Relevant in PAD Formation
3.1. Aortic Cells and Dysfunction
3.2. Atherosclerosis
3.3. Inflammation
3.4. Arterial Stiffness and Calcification
3.5. Vitamin D Status and Angiogenesis
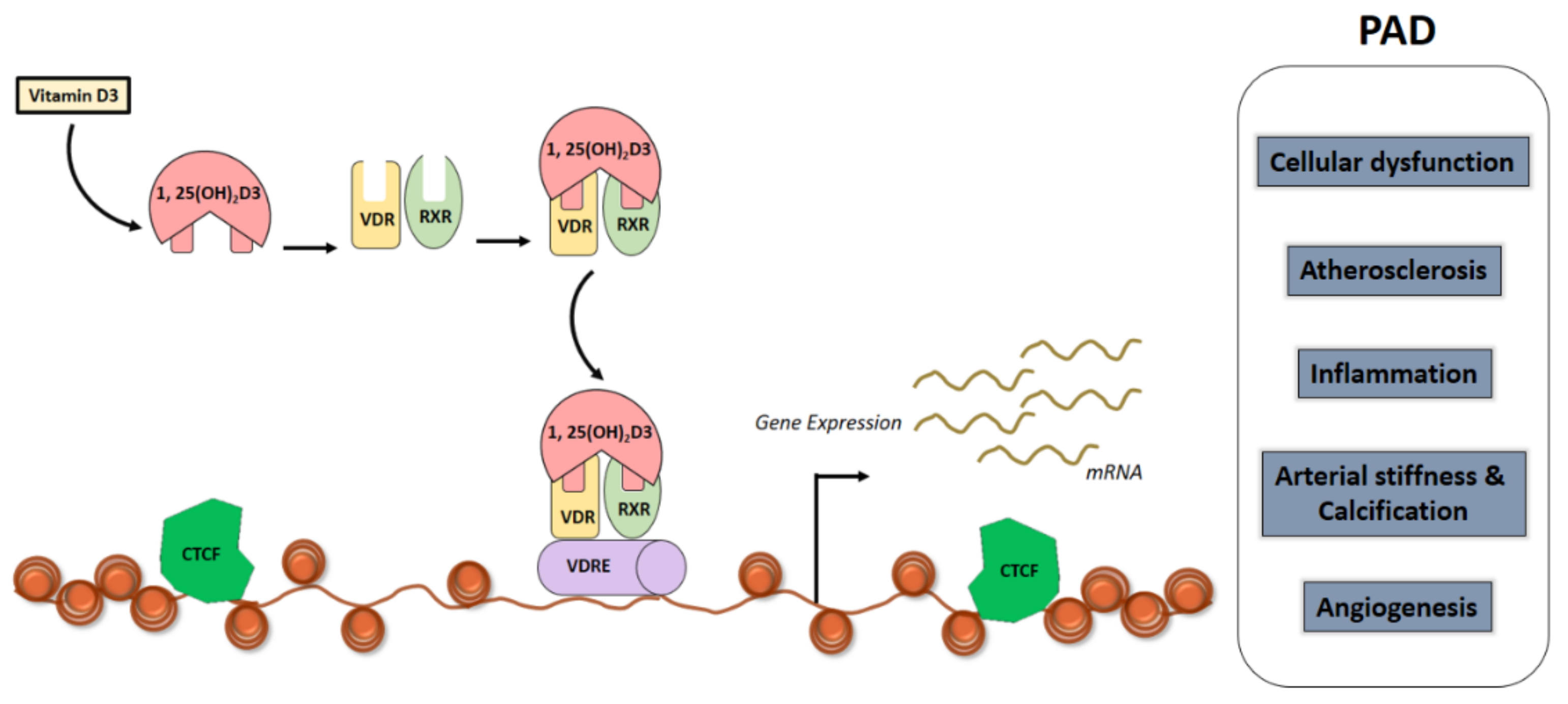
4. Vitamin D and the Genome
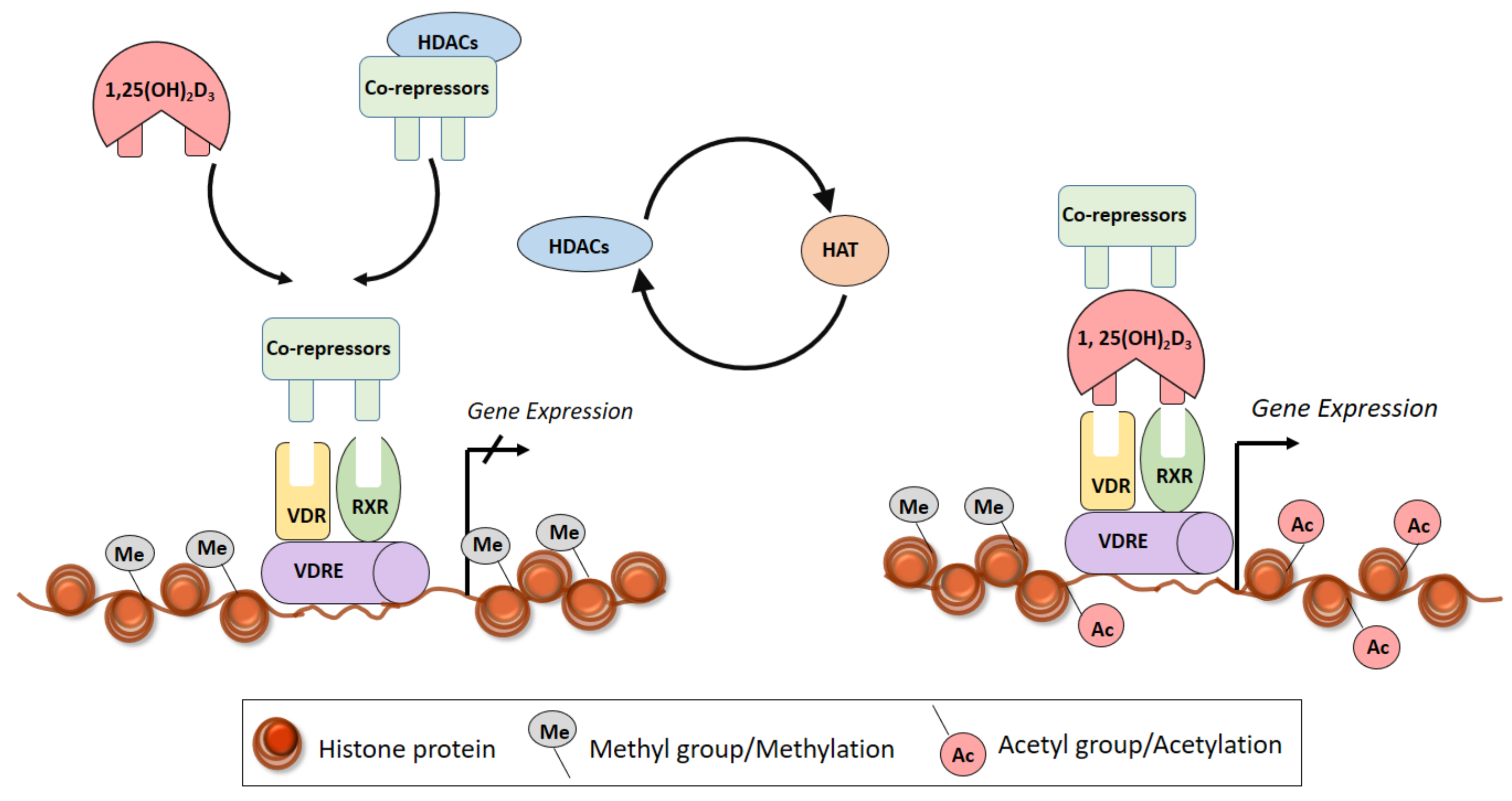
5. Vitamin D and the Epigenome
5.1. Histone Modifications
5.2. DNA Methylation
6. Future Direction
Funding
Acknowledgments
Abbreviation
| AAA | Abdominal aortic aneurysm |
| AngII | Angiotensin II |
| ARIC | Atherosclerosis Risk In Communities |
| CLI | Critical limb ischemia |
| CVD | Cardio vascular disease |
| DBP | Vitamin D binding protein |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| eNOS | Endothelial nitric oxide synthase |
| EPC | Endothelial progenitor cell |
| HAT | Histone acetyl transferase |
| HDAC | Histone deacetylase |
| IC | Intermittent claudication |
| MMP | Matrix metalloproteinase |
| NO | Nitric oxide |
| (OH)D | Hydroxyvitamin D |
| PAD | Peripheral arterial disease |
| PAOD | Peripheral arterial occlusive disease |
| PBMNC | Peripheral blood mononuclear cells |
| RAAS | Renin-angiotensin-aldosterone system |
| RXR | Retinoid X receptor |
| TSS | Transcription start site |
| VDR | Vitamin D receptor |
| VSMC | Vascular smooth muscle cell |
References
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A critical and essential micronutrient for human health. Front. Physiol. 2014, 5, 248. [Google Scholar] [CrossRef]
- Hilger, J.; Friedel, A.; Herr, R.; Rausch, T.; Roos, F.; Wahl, D.A.; Pierroz, D.D.; Weber, P.; Hoffmann, K. A systematic review of vitamin D status in populations worldwide. Br. J. Nutr. 2014, 111, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Pludowski, P.; Grant, W.B.; Bhattoa, H.P.; Bayer, M.; Povoroznyuk, V.; Rudenka, E.; Ramanau, H.; Varbiro, S.; Rudenka, A.; Karczmarewicz, E.; et al. Vitamin d status in central Europe. Int. J. Endocrinol. 2014, 2014, 589587. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Schleithoff, S.S.; Koerfer, R. Putting cardiovascular disease and vitamin D insufficiency into perspective. Br. J. Nutr. 2005, 94, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef]
- Holick, M.F.; Chen, T.C. Vitamin D deficiency: A worldwide problem with health consequences. Am. J. Clin. Nutr. 2008, 87, 1080s–1086s. [Google Scholar] [CrossRef]
- Wang, L.; Ma, J.; Manson, J.E.; Buring, J.E.; Gaziano, J.M.; Sesso, H.D. A prospective study of plasma vitamin D metabolites, vitamin D receptor gene polymorphisms, and risk of hypertension in men. Eur. J. Nutr. 2013, 52, 1771–1779. [Google Scholar] [CrossRef]
- Girgis, C.M.; Clifton-Bligh, R.J.; Turner, N.; Lau, S.L.; Gunton, J.E. Effects of vitamin D in skeletal muscle: Falls, strength, athletic performance and insulin sensitivity. Clin. Endocrinol. 2014, 80, 169–181. [Google Scholar] [CrossRef]
- Van de Luijtgaarden, K.M.; Voute, M.T.; Hoeks, S.E.; Bakker, E.J.; Chonchol, M.; Stolker, R.J.; Rouwet, E.V.; Verhagen, H.J. Vitamin D deficiency may be an independent risk factor for arterial disease. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 301–306. [Google Scholar] [CrossRef]
- Golledge, J. Lower-limb arterial disease. Lancet. 1997, 350, 1459–1465. [Google Scholar] [CrossRef]
- Cooke, J.P.; Chen, Z. A compendium on peripheral arterial disease. Circ. Res. 2015, 116, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Duff, S.; Mafilios, M.S.; Bhounsule, P.; Hasegawa, J.T. The burden of critical limb ischemia: a review of recent literature. Vasc. Health Risk Manag. 2019, 15, 187–208. [Google Scholar] [CrossRef] [PubMed]
- Olin, J.W.; Sealove, B.A. Peripheral artery disease: Current insight into the disease and its diagnosis and management. Mayo Clin. Proc. 2010, 85, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet. 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Moxon, J.V.; Parr, A.; Emeto, T.I.; Walker, P.; Norman, P.E.; Golledge, J. Diagnosis and monitoring of abdominal aortic aneurysm: Current status and future prospects. Curr. Probl. Cardiol. 2010, 35, 512–548. [Google Scholar] [CrossRef]
- Sampson, U.K.A.; Norman, P.E.; Fowkes, F.G.R.; Aboyans, V.; Song, Y.; Harrell, F.E., Jr.; Forouzanfar, M.H.; Naghavi, M.; Denenberg, J.O.; McDermott, M.M.; et al. Estimation of Global and Regional Incidence and Prevalence of Abdominal Aortic Aneurysms 1990 to 2010. Glob. Heart. 2014, 9, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Aboyans, V. Epidemiology of peripheral artery disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef]
- Weinberg, M.D.; Lau, J.F.; Rosenfield, K.; Olin, J.W. Peripheral artery disease. Part 2: Medical and endovascular treatment. Nat. Rev. Cardiol. 2011, 8, 429–441. [Google Scholar] [CrossRef]
- Ankle Brachial Index, Collaboration; Fowkes, F.G.; Murray, G.D.; Butcher, I.; Heald, C.L.; Lee, R.J.; Chambless, L.E.; Folsom, A.R.; Hirsch, A.T.; Dramaix, M.; et al. Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: A meta-analysis. JAMA 2008, 300, 197–208. [Google Scholar] [PubMed]
- Ramirez, J.L.; Drudi, L.M.; Grenon, S.M. Review of biologic and behavioral risk factors linking depression and peripheral artery disease. Vasc. Med. 2018, 23, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Rapson, I.R.; Michos, E.D.; Alonso, A.; Hirsch, A.T.; Matsushita, K.; Reis, J.P.; Lutsey, P.L. Serum 25-hydroxyvitamin D is associated with incident peripheral artery disease among white and black adults in the ARIC study cohort. Atherosclerosis 2017, 257, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amer, M.; Narotsky, D.L.; Qayyum, R. 25-Hydroxyvitamin D and ankle-brachial blood pressure index in adults without peripheral artery disease. Clin. Transl. Sci. 2014, 7, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Stricker, H.; Tosi Bianda, F.; Guidicelli-Nicolosi, S.; Limoni, C.; Colucci, G. Effect of a single, oral, high-dose vitamin D supplementation on endothelial function in patients with peripheral arterial disease: A randomised controlled pilot study. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, V.C.; Bailey, B.A.; Kuriacose, R.; Copeland, R.J.; Manning, T.; Peiris, A.N. The relationship of vitamin D status to cardiovascular risk factors and amputation risk in veterans with peripheral arterial disease. J. Am. Med. Dir. Assoc. 2011, 12, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Zagura, M.; Serg, M.; Kampus, P.; Zilmer, M.; Eha, J.; Unt, E.; Lieberg, J.; Cockcroft, J.R.; Kals, J. Aortic stiffness and vitamin D are independent markers of aortic calcification in patients with peripheral arterial disease and in healthy subjects. Eur. J. Vasc. Endovasc. Surg. 2011, 42, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Muntner, P.; Michos, E.D.; Uribarri, J.; Weber, C.; Sharma, J.; Raggi, P. Serum 25-hydroxyvitamin D levels and the prevalence of peripheral arterial disease: Results from NHANES 2001 to 2004. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1179–1185. [Google Scholar] [CrossRef]
- Reis, J.P.; Michos, E.D.; Von Muhlen, D.; Miller, E.R., 3rd. Differences in vitamin D status as a possible contributor to the racial disparity in peripheral arterial disease. Am. J. Clin. Nutr. 2008, 88, 1469–1477. [Google Scholar] [CrossRef] [Green Version]
- Fahrleitner-Pammer, A.; Obernosterer, A.; Pilger, E.; Dobnig, H.; Dimai, H.P.; Leb, G.; Kudlacek, S.; Obermayer-Pietsch, B.M. Hypovitaminosis D, impaired bone turnover and low bone mass are common in patients with peripheral arterial disease. Osteoporos. Int. 2005, 16, 319–324. [Google Scholar] [CrossRef]
- Fahrleitner, A.; Dobnig, H.; Obernosterer, A.; Pilger, E.; Leb, G.; Weber, K.; Kudlacek, S.; Obermayer-Pietsch, B.M. Vitamin D deficiency and secondary hyperparathyroidism are common complications in patients with peripheral arterial disease. J. Gen. Intern. Med. 2002, 17, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, J.Y.; Sasha, S.R.; Ngu, P.J.; Warren, J.L.; Wark, J.; Dart, A.M.; Shaw, J.A. Circulating vitamin D levels are associated with the presence and severity of coronary artery disease but not peripheral arterial disease in patients undergoing coronary angiography. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 274–279. [Google Scholar] [CrossRef]
- Veronese, N.; De Rui, M.; Bolzetta, F.; Toffanello, E.D.; Coin, A.; Zambon, S.; Corti, M.C.; Baggio, G.; Perissinotto, E.; Maggi, S.; et al. Serum 25-Hydroxyvitamin D and the Incidence of Peripheral Artery Disease in the Elderly: The Pro.V.A Study. J. Atheroscler. Thromb. 2015, 22, 726–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.M.; Liu, K.; Ferrucci, L.; Tian, L.; Guralnik, J.; Kopp, P.; Tao, H.; Van Horn, L.; Liao, Y.; Green, D.; et al. Vitamin D status and functional performance in peripheral artery disease. Vasc. Med. 2012, 17, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazidi, M.; Wong, N.D.; Katsiki, N.; Mikhailidis, D.P.; Banach, M. Dietary patterns, plasma vitamins and trans fatty acids are associated with peripheral artery disease. Lipids Health Dis. 2017, 16, 254. [Google Scholar] [CrossRef] [PubMed]
- Nsengiyumva, V.; Fernando, M.E.; Moxon, J.V.; Krishna, S.M.; Pinchbeck, J.; Omer, S.M.; Morris, D.R.; Jones, R.E.; Moran, C.S.; Seto, S.W.; et al. The association of circulating 25-hydroxyvitamin D concentration with peripheral arterial disease: A meta-analysis of observational studies. Atherosclerosis 2015, 243, 645–651. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Liu, K.; Ferrucci, L.; Tian, L.; Guralnik, J.; Kopp, P.; Van Horn, L.; Liao, Y.; Green, D.; Kibbe, M.; et al. Vitamin D status, functional decline, and mortality in peripheral artery disease. Vasc. Med. 2014, 19, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Veldurthy, V.; Wei, R.; Oz, L.; Dhawan, P.; Jeon, Y.H.; Christakos, S. Vitamin D, calcium homeostasis and aging. Bone Res. 2016, 4, 16041. [Google Scholar] [CrossRef]
- Monaro, S.; West, S.; Gullick, J. An integrative review of health-related quality of life in patients with critical limb ischaemia. J Clin. Nurs. 2017, 26((19–20)), 2826–2844. [Google Scholar] [CrossRef]
- Van Ballegooijen, A.J.; Reinders, I.; Visser, M.; Brouwer, I.A. Parathyroid hormone and cardiovascular disease events: A systematic review and meta-analysis of prospective studies. Am. Heart J. 2013, 165, 655–664. [Google Scholar] [CrossRef]
- Gamberi, T.; Puglia, M.; Guidi, F.; Magherini, F.; Bini, L.; Marzocchini, R.; Modesti, A.; Modesti, P.A. A proteomic approach to identify plasma proteins in patients with abdominal aortic aneurysm. Mol. Biosyst. 2011, 7, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- United States Census Bureau, Population Division. Annual Estimates of the Resident Population by Single Year of Age and Sex for the United States: April 1, 2010 to July 1, 2017. Release date June 2018.
- Wanhainen, A.; Bjorck, M.; Boman, K.; Rutegard, J.; Bergqvist, D. Influence of diagnostic criteria on the prevalence of abdominal aortic aneurysm. J. Vasc. Surg. 2001, 34, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultgren, R.; Forsberg, J.; Alfredsson, L.; Swedenborg, J.; Leander, K. Regional variation in the incidence of abdominal aortic aneurysm in Sweden. Br. J. Surg. 2012, 99, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Uyan, U.; Melek, M. The relationship between vitamin D deficiency and thoracic aortic dilatation. Vasa. 2012, 41, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.Y.; Flicker, L.; Yeap, B.B.; McCaul, K.A.; Hankey, G.J.; Norman, P.E. Is hypovitaminosis D associated with abdominal aortic aneurysm, and is there a dose-response relationship? Eur. J. Vasc. Endovasc. Surg. 2013, 45, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Ponda, M.P.; Huang, X.; Odeh, M.A.; Breslow, J.L.; Kaufman, H.W. Vitamin D may not improve lipid levels: A serial clinical laboratory data study. Circulation 2012, 126, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Ford, E.S.; Li, C.; Croft, J.B. Serum 25-hydroxyvitamin D levels and all-cause and cardiovascular disease mortality among US adults with hypertension: The NHANES linked mortality study. J. Hypertens. 2012, 30, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Umemoto, T. Vitamins and abdominal aortic aneurysm. Int. Angiol. 2017, 36, 21–30. [Google Scholar]
- Spadaccio, C.; Coccia, R.; Perluigi, M.; Pupo, G.; Schinina, M.E.; Giorgi, A.; Blarzino, C.; Nappi, F.; Sutherland, F.W.; Chello, M.; et al. Redox proteomic analysis of serum from aortic anerurysm patients: Insights on oxidation of specific protein target. Mol. Biosyst. 2016, 12, 2168–2177. [Google Scholar] [CrossRef]
- Nieuwland, A.J.; Kokje, V.B.; Koning, O.H.; Hamming, J.F.; Szuhai, K.; Claas, F.H.; Lindeman, J.H. Activation of the vitamin D receptor selectively interferes with calcineurin-mediated inflammation: A clinical evaluation in the abdominal aortic aneurysm. Lab. Investig. 2016, 96, 784–790. [Google Scholar] [CrossRef]
- Lutsey, P.L.; Rooney, M.R.; Folsom, A.R.; Michos, E.D.; Alonso, A.; Tang, W. Markers of vitamin D metabolism and incidence of clinically diagnosed abdominal aortic aneurysm: The Atherosclerosis Risk in Communities Study. Vasc. Med. 2018, 23, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Urbonavicius, S.; Lindholt, J.S.; Delbosc, S.; Urbonaviciene, G.; Henneberg, E.W.; Vorum, H.; Meilhac, O.; Honore, B. Proteins associated with the size and expansion rate of the abdominal aortic aneurysm wall as identified by proteomic analysis. Interact. Cardiovasc. Thorac. Surg. 2010, 11, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Chun, R.F.; Peercy, B.E.; Orwoll, E.S.; Nielson, C.M.; Adams, J.S.; Hewison, M. Vitamin D and DBP: The free hormone hypothesis revisited. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 132–137. [Google Scholar] [CrossRef]
- Norman, P.E.; Powell, J.T. Vitamin D, shedding light on the development of disease in peripheral arteries. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Martorell, S.; Hueso, L.; Gonzalez-Navarro, H.; Collado, A.; Sanz, M.J.; Piqueras, L. Vitamin D Receptor Activation Reduces Angiotensin-II-Induced Dissecting Abdominal Aortic Aneurysm in Apolipoprotein E-Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1587–1597. [Google Scholar] [CrossRef]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Li, Y.C. Vitamin D regulation of the renin-angiotensin system. J. Cell Biochem. 2003, 88, 327–331. [Google Scholar] [CrossRef]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef]
- Garcia, I.M.; Altamirano, L.; Mazzei, L.; Fornes, M.; Cuello-Carrion, F.D.; Ferder, L.; Manucha, W. Vitamin D receptor-modulated Hsp70/AT1 expression may protect the kidneys of SHRs at the structural and functional levels. Cell Stress Chaperones. 2014, 19, 479–491. [Google Scholar] [CrossRef]
- Dong, J.; Wong, S.L.; Lau, C.W.; Lee, H.K.; Ng, C.F.; Zhang, L.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Huang, Y. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. Eur. Heart J. 2012, 33, 2980–2990. [Google Scholar] [CrossRef] [Green Version]
- Kassi, E.; Adamopoulos, C.; Basdra, E.K.; Papavassiliou, A.G. Role of vitamin D in atherosclerosis. Circulation 2013, 128, 2517–2531. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.E.; Powell, J.T. Vitamin D and Cardiovascular Disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Brewer, L.C.; Michos, E.D.; Reis, J.P. Vitamin D in atherosclerosis, vascular disease, and endothelial function. Curr. Drug Targets. 2011, 12, 54–60. [Google Scholar] [CrossRef]
- Somjen, D.; Weisman, Y.; Kohen, F.; Gayer, B.; Limor, R.; Sharon, O.; Jaccard, N.; Knoll, E.; Stern, N. 25-hydroxyvitamin D3-1alpha-hydroxylase is expressed in human vascular smooth muscle cells and is upregulated by parathyroid hormone and estrogenic compounds. Circulation 2005, 111, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Merke, J.; Milde, P.; Lewicka, S.; Hugel, U.; Klaus, G.; Mangelsdorf, D.J.; Haussler, M.R.; Rauterberg, E.W.; Ritz, E. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J. Clin. Investig. 1989, 83, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Morris, R.C., Jr.; Ives, H.E. 1,25-dihydroxyvitamin D3 modulates growth of vascular smooth muscle cells. J. Clin. Investig. 1991, 87, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ichiyama, T.; Ohsaki, A.; Hasegawa, S.; Shiraishi, M.; Furukawa, S. Anti-inflammatory effect of 1alpha,25-dihydroxyvitamin D(3) in human coronary arterial endothelial cells: Implication for the treatment of Kawasaki disease. J. Steroid Biochem. Mol. Biol. 2009, 113, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Cardus, A.; Parisi, E.; Gallego, C.; Aldea, M.; Fernandez, E.; Valdivielso, J.M. 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006, 69, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Miguel, P.; Valdivielso, J.M.; Medrano-Andres, D.; Roman-Garcia, P.; Cano-Penalver, J.L.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Lopez-Ongil, S. The active form of vitamin D, calcitriol, induces a complex dual upregulation of endothelin and nitric oxide in cultured endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1085–E1096. [Google Scholar] [CrossRef] [Green Version]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-dihydroxyvitamin D(3) by human endothelial cells is regulated by inflammatory cytokines: A novel autocrine determinant of vascular cell adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar]
- Riek, A.E.; Oh, J.; Sprague, J.E.; Timpson, A.; De las Fuentes, L.; Bernal-Mizrachi, L.; Schechtman, K.B.; Bernal-Mizrachi, C. Vitamin D suppression of endoplasmic reticulum stress promotes an antiatherogenic monocyte/macrophage phenotype in type 2 diabetic patients. J. Biol. Chem. 2012, 287, 38482–38494. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.J.; Jafri, M.; Wehmeier, K.R.; Onstead-Haas, L.M.; Mooradian, A.D. Inhibition of endoplasmic reticulum stress and oxidative stress by vitamin D in endothelial cells. Free Radic. Biol. Med. 2016, 99, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Uberti, F.; Grossini, E.; Vacca, G.; Carda, S.; Invernizzi, M.; Cisari, C. 1alpha,25-dihydroxycholecalciferol induces nitric oxide production in cultured endothelial cells. Cell Physiol. Biochem. 2011, 27, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Aihara, K.; Azuma, H.; Akaike, M.; Ikeda, Y.; Yamashita, M.; Sudo, T.; Hayashi, H.; Yamada, Y.; Endoh, F.; Fujimura, M.; et al. Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J. Biol. Chem. 2004, 279, 35798–35802. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Elenkova, M.; Tipton, D.A.; Karydis, A.; Stein, S.H. Vitamin D attenuates human gingival fibroblast inflammatory cytokine production following advanced glycation end product interaction with receptors for AGE. J. Periodontal Res. 2019, 54, 154–163. [Google Scholar] [CrossRef]
- Dziedzic, E.A.; Gasior, J.S.; Pawlowski, M.; Wodejko-Kucharska, B.; Saniewski, T.; Marcisz, A.; Dabrowski, M.J. Vitamin D level is associated with severity of coronary artery atherosclerosis and incidence of acute coronary syndromes in non-diabetic cardiac patients. Arch. Med. Sci. 2019, 15, 359–368. [Google Scholar] [CrossRef]
- Oh, J.; Weng, S.; Felton, S.K.; Bhandare, S.; Riek, A.; Butler, B.; Proctor, B.M.; Petty, M.; Chen, Z.; Schechtman, K.B. 1, 25 (OH) 2 vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation 2009, 120, 687–698. [Google Scholar] [CrossRef]
- Timms, P.M.; Mannan, N.; Hitman, G.A.; Noonan, K.; Mills, P.G.; Syndercombe-Court, D.; Aganna, E.; Price, C.P.; Boucher, B.J. Circulating MMP9, vitamin D and variation in the TIMP-1 response with VDR genotype: Mechanisms for inflammatory damage in chronic disorders? QJM 2002, 95, 787–796. [Google Scholar] [CrossRef]
- Kalkan, G.Y.; Gur, M.; Koyunsever, N.Y.; Seker, T.; Gozukara, M.Y.; Ucar, H.; Kaypakli, O.; Baykan, A.O.; Akyol, S.; Turkoglu, C.; et al. Serum 25-Hydroxyvitamin D Level and Aortic Intima-Media Thickness in Patients without Clinical Manifestation of Atherosclerotic Cardiovascular Disease. J. Clin. Lab. Anal. 2014, 29, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Chua, G.T.; Chan, Y.C.; Cheng, S.W. Vitamin D status and peripheral arterial disease: Evidence so far. Vasc. Health Risk Manag. 2011, 7, 671–675. [Google Scholar] [PubMed]
- Martins, D.; Wolf, M.; Pan, D.; Zadshir, A.; Tareen, N.; Thadhani, R.; Felsenfeld, A.; Levine, B.; Mehrotra, R.; Norris, K. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: Data from the Third National Health and Nutrition Examination Survey. Arch. Intern. Med. 2007, 167, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Quigley, F.; Velu, R.; Walker, P.J.; Moxon, J.V. Association of impaired fasting glucose, diabetes and their management with the presentation and outcome of peripheral artery disease: A cohort study. Cardiovasc. Diabetol. 2014, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Yamashita, T.; Sasaki, N.; Nakajima, K.; Kita, T.; Shinohara, M.; Ishida, T.; Hirata, K. Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Cutolo, M. Vitamin D and autoimmune rheumatic diseases. Rheumatology 2009, 48, 210–212. [Google Scholar] [CrossRef]
- Lee, J.H.; O’Keefe, J.H.; Bell, D.; Hensrud, D.D.; Holick, M.F. Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor? J. Am. Coll. Cardiol. 2008, 52, 1949–1956. [Google Scholar] [CrossRef]
- Kunadian, V.; Ford, G.A.; Bawamia, B.; Qiu, W.; Manson, J.E. Vitamin D deficiency and coronary artery disease: A review of the evidence. Am. Heart J. 2014, 167, 283–291. [Google Scholar] [CrossRef]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Lemire, J.M.; Adams, J.S.; Kermani-Arab, V.; Bakke, A.C.; Sakai, R.; Jordan, S.C. 1,25-Dihydroxyvitamin D3 suppresses human T helper/inducer lymphocyte activity in vitro. J. Immunol. 1985, 134, 3032–3035. [Google Scholar] [PubMed]
- Jablonski, K.L.; Chonchol, M.; Pierce, G.L.; Walker, A.E.; Seals, D.R. 25-Hydroxyvitamin D deficiency is associated with inflammation-linked vascular endothelial dysfunction in middle-aged and older adults. Hypertension 2011, 57, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Laird, E.; McNulty, H.; Ward, M.; Hoey, L.; McSorley, E.; Wallace, J.; Carson, E.; Molloy, A.; Healy, M.; Casey, M. Vitamin D deficiency is associated with inflammation in older Irish adults. J. Clin. Endocrinol. Metab. 2014, 99, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.B.; Gysemans, C.A.; Demengeot, J.; Da Cunha, J.P.; Vanherwegen, A.S.; Overbergh, L.; Van Belle, T.L.; Pauwels, F.; Verstuyf, A.; Korf, H.; et al. 1,25-Dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J. Immunol. 2014, 192, 4210–4220. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Hu, Z.L.; He, X.J.; Dang, X.Q. Intravenous transfusion of endothelial progenitor cells that overexpress vitamin D receptor inhibits atherosclerosis in apoE-deficient mice. Biomed Pharmacother. 2016, 84, 1233–1242. [Google Scholar] [CrossRef]
- Müller, K.; Haahr, P.; Diamant, M.; Rieneck, K.; Kharazmi, A.; Bendtzen, K. 1, 25-dihydroxyvitamin D 3 inhibits cytokine production by human blood monocytes at the post-transcriptional level. Cytokine 1992, 4, 506–512. [Google Scholar] [CrossRef]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Ronnemaa, T.; Laakso, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef]
- Reiss, A.B.; Miyawaki, N.; Moon, J.; Kasselman, L.J.; Voloshyna, I.; D’Avino, R., Jr.; De Leon, J. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis 2018, 278, 49–59. [Google Scholar] [CrossRef]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef]
- Dong, Y.; Stallmann-Jorgensen, I.S.; Pollock, N.K.; Harris, R.A.; Keeton, D.; Huang, Y.; Li, K.; Bassali, R.; Guo, D.H.; Thomas, J.; et al. A 16-week randomized clinical trial of 2000 international units daily vitamin D3 supplementation in black youth: 25-hydroxyvitamin D, adiposity, and arterial stiffness. J. Clin. Endocrinol. Metab. 2010, 95, 4584–4591. [Google Scholar] [CrossRef]
- Kumar, V.; Yadav, A.K.; Lal, A.; Kumar, V.; Singhal, M.; Billot, L.; Gupta, K.L.; Banerjee, D.; Jha, V. A Randomized Trial of Vitamin D Supplementation on Vascular Function in CKD. J. Am. Soc. Nephrol. 2017, 28, 3100–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Sun, Y.; Agrawal, D.K. Vitamin D deficiency and essential hypertension. J. Am. Soc. Hypertens. 2015, 9, 885–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, S.; Lund, R.J.; Chaudhary, L.R.; Geurs, T.; Hruska, K.A. Vitamin D receptor activators can protect against vascular calcification. J. Am. Soc. Nephrol. 2008, 19, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Haidar, M.; Placzko, S.; Niendorf, R.; Darashchonak, N.; Hubel, C.A.; Von Versen-Hoynck, F. Vitamin D improves the angiogenic properties of endothelial progenitor cells. Am. J. Physiol. Cell Physiol. 2012, 303, C954–C962. [Google Scholar] [CrossRef] [PubMed]
- Arfian, N.; Kusuma, M.H.; Anggorowati, N.; Nugroho, D.B.; Jeffilano, A.; Suzuki, Y.; Ikeda, K.; Emoto, N. Vitamin D upregulates endothelin-1, ETBR, eNOS mRNA expression and attenuates vascular remodelling and ischemia in kidney fibrosis model in mice. Physiol. Res. 2018, 67 (Suppl. 1), S137–S147. [Google Scholar] [CrossRef]
- Shokravi, M.T.; Marcus, D.M.; Alroy, J.; Egan, K.; Saornil, M.A.; Albert, D.M. Vitamin D inhibits angiogenesis in transgenic murine retinoblastoma. Investig. Ophthalmol. Vis. Sci. 1995, 36, 83–87. [Google Scholar]
- Osborne, J.E.; Hutchinson, P.E. Vitamin D and systemic cancer: Is this relevant to malignant melanoma? Br. J. Dermatol. 2002, 147, 197–213. [Google Scholar] [CrossRef]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef]
- Albert, D.M.; Scheef, E.A.; Wang, S.; Mehraein, F.; Darjatmoko, S.R.; Sorenson, C.M.; Sheibani, N. Calcitriol is a potent inhibitor of retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2327–2334. [Google Scholar] [CrossRef]
- Jamali, N.; Wang, S.; Darjatmoko, S.R.; Sorenson, C.M.; Sheibani, N. Vitamin D receptor expression is essential during retinal vascular development and attenuation of neovascularization by 1, 25(OH)2D3. PLoS ONE 2017, 12, e0190131. [Google Scholar] [CrossRef]
- Trujillo, V.; Marin-Luevano, P.; Gonzalez-Curiel, I.; Rodriguez-Carlos, A.; Ramirez-Reyes, M.; Layseca-Espinosa, E.; Enciso-Moreno, J.A.; Diaz, L.; Rivas-Santiago, B. Calcitriol promotes proangiogenic molecules in keratinocytes in a diabetic foot ulcer model. J. Steroid Biochem. Mol. Biol. 2017, 174, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.M.; Zhang, R.; Park, C.; Nong, Z.; Yin, H.; Wilson, R.B.; Sutherland, B.G.; Sawyez, C.G.; Pickering, J.G.; Borradaile, N.M. Vitamin D intervention does not improve vascular regeneration in diet-induced obese male mice with peripheral ischemia. J. Nutr. Biochem. 2019, 70, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.B.; Levine, M.A.; Bell, N.H.; Mangelsdorf, D.J.; Russell, D.W. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc. Natl. Acad. Sci. USA 2004, 101, 7711–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Zhang, F.; Richards, J.B.; Kestenbaum, B.; Van Meurs, J.B.; Berry, D.; Kiel, D.P.; Streeten, E.A.; Ohlsson, C.; Koller, D.L.; et al. Common genetic determinants of vitamin D insufficiency: A genome–wide association study. Lancet. 2010, 376, 180–188. [Google Scholar] [CrossRef]
- Ahn, J.; Yu, K.; Stolzenberg-Solomon, R.; Simon, K.C.; McCullough, M.L.; Gallicchio, L.; Jacobs, E.J.; Ascherio, A.; Helzlsouer, K.; Jacobs, K.B.; et al. Genome–wide association study of circulating vitamin D levels. Hum. Mol. Genet. 2010, 19, 2739–2745. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B. The Vitamin D Receptor: New Paradigms for the Regulation of Gene Expression by 1,25-Dihydroxyvitamin D(3). Endocrinol. Metab. Clin. North. Am. 2010, 39, 255–269. [Google Scholar] [CrossRef]
- King, A.N.; Beer, D.G.; Christensen, P.J.; Simpson, R.U.; Ramnath, N. The vitamin D/CYP24A1 story in cancer. Anticancer Agents Med. Chem. 2010, 10, 213–224. [Google Scholar] [CrossRef]
- Wang, T.J. Vitamin D and Cardiovascular Disease. Annu. Rev. Med. 2016, 67, 261–272. [Google Scholar] [CrossRef]
- Trump, D.L.; Hershberger, P.A.; Bernardi, R.J.; Ahmed, S.; Muindi, J.; Fakih, M.; Yu, W.D.; Johnson, C.S. Anti–tumor activity of calcitriol: Pre–clinical and clinical studies. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 519–526. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B. Fundamentals of vitamin D hormone-regulated gene expression. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 5–11. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; Van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Seuter, S.; De Mello, V.D.; Schwab, U.; Voutilainen, S.; Pulkki, K.; Nurmi, T.; Virtanen, J.; Tuomainen, T.P.; Uusitupa, M. Primary vitamin D target genes allow a categorization of possible benefits of vitamin D(3) supplementation. PLoS ONE 2013, 8, e71042. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Spreading of silent chromatin: Inaction at a distance. Nat. Rev. Genet. 2006, 7, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Trollope, A.F.; Golledge, J. The relevance of epigenetics to occlusive cerebral and peripheral arterial disease. Clin. Sci. 2015, 128, 537–558. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Moxon, J.V.; Golledge, J. A Review of the Pathophysiology and Potential Biomarkers for Peripheral Artery Disease. Int. J. Mol. Sci. 2015, 16, 11294–11322. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.M.; Seto, S.W.; Jose, R.J.; Li, J.; Morton, S.K.; Biros, E.; Wang, Y.; Nsengiyumva, V.; Lindeman, J.H.; Loots, G.G.; et al. Wnt Signaling Pathway Inhibitor Sclerostin Inhibits Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 37, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.S.; Biros, E.; Krishna, S.M.; Wang, Y.; Tikellis, C.; Morton, S.K.; Moxon, J.V.; Cooper, M.E.; Norman, P.E.; Burrell, L.M.; et al. Resveratrol Inhibits Growth of Experimental Abdominal Aortic Aneurysm Associated With Upregulation of Angiotensin-Converting Enzyme 2. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2195–2203. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Benkusky, N.A.; Pike, J.W. 1,25-Dihydroxyvitamin D3 induced histone profiles guide discovery of VDR action sites. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 19–21. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer. 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Seuter, S. Dynamics of nuclear receptor target gene regulation. Chromosoma 2010, 119, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rosa, M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Vitamin D3: A helpful immuno-modulator. Immunology. 2011, 134, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Seuter, S.; Pehkonen, P.; Heikkinen, S.; Carlberg, C. Dynamics of 1alpha,25-dihydroxyvitamin D3-dependent chromatin accessibility of early vitamin D receptor target genes. Biochim. Biophys. Acta 2013, 1829, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shevde, N.K.; Pike, J.W. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J. Bone Miner. Res. 2005, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Khanim, F.L.; Gommersall, L.M.; Wood, V.H.; Smith, K.L.; Montalvo, L.; O’Neill, L.P.; Xu, Y.; Peehl, D.M.; Stewart, P.M.; Turner, B.M.; et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 2004, 23, 6712–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, S.F.; Moore, J.S.; Walker, E.; Driver, P.M.; Engel, J.; Edwards, C.E.; Brown, G.; Uskokovic, M.R.; Campbell, M.J. Synergistic growth inhibition of prostate cancer cells by 1 alpha,25 Dihydroxyvitamin D(3) and its 19-nor-hexafluoride analogs in combination with either sodium butyrate or trichostatin A. Oncogene 2001, 20, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Polly, P.; Herdick, M.; Moehren, U.; Baniahmad, A.; Heinzel, T.; Carlberg, C. VDR-Alien: A novel, DNA-selective vitamin D(3) receptor-corepressor partnership. FASEB J. 2000, 14, 1455–1463. [Google Scholar] [CrossRef]
- Banwell, C.M.; O’Neill, L.P.; Uskokovic, M.R.; Campbell, M.J. Targeting 1alpha,25-dihydroxyvitamin D3 antiproliferative insensitivity in breast cancer cells by co-treatment with histone deacetylation inhibitors. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenet. 2011, 2, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golledge, J.; Biros, E.; Bingley, J.; Iyer, V.; Krishna, S.M. Epigenetics and Peripheral Artery Disease. Curr. Atheroscler. Rep. 2016, 18, 15. [Google Scholar] [CrossRef]
- Aranow, C. Vitamin D and the immune system. J. Investig. Med. 2011, 59, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Molnar, F. Vitamin D receptor signaling and its therapeutic implications: Genome-wide and structural view. Can. J. Physiol. Pharm. 2015, 93, 311–318. [Google Scholar] [CrossRef]
- Oma, I.; Olstad, O.K.; Andersen, J.K.; Lyberg, T.; Molberg, O.; Fostad, I.; Wang Fagerland, M.; Almdahl, S.M.; Rynning, S.E. Differential expression of vitamin D associated genes in the aorta of coronary artery disease patients with and without rheumatoid arthritis. PLoS ONE 2018, 13, e0202346. [Google Scholar] [CrossRef] [PubMed]
- Bremmer, F.; Thelen, P.; Pottek, T.; Behnes, C.L.; Radzun, H.J.; Schweyer, S. Expression and function of the vitamin D receptor in malignant germ cell tumour of the testis. Anticancer Res. 2012, 32, 341–349. [Google Scholar]
- Hossein-nezhad, A.; Spira, A.; Holick, M.F. Influence of vitamin D status and vitamin D3 supplementation on genome wide expression of white blood cells: A randomized double-blind clinical trial. PLoS ONE 2013, 8, e58725. [Google Scholar] [CrossRef]
- Carlberg, C.; Seuter, S.; Nurmi, T.; Tuomainen, T.P.; Virtanen, J.K.; Neme, A. In vivo response of the human epigenome to vitamin D: A Proof-of-principle study. J. Steroid Biochem. Mol. Biol. 2018, 180, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seuter, S.; Virtanen, J.K.; Nurmi, T.; Pihlajamaki, J.; Mursu, J.; Voutilainen, S.; Tuomainen, T.P.; Neme, A.; Carlberg, C. Molecular evaluation of vitamin D responsiveness of healthy young adults. J. Steroid Biochem. Mol. Biol. 2017, 174, 314–321. [Google Scholar] [CrossRef]
- Neme, A.; Seuter, S.; Malinen, M.; Nurmi, T.; Tuomainen, T.P.; Virtanen, J.K.; Carlberg, C. In vivo transcriptome changes of human white blood cells in response to vitamin D. J. Steroid Biochem. Mol. Biol. 2019, 188, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Seuter, S.; Neme, A.; Carlberg, C. Epigenome-wide effects of vitamin D and their impact on the transcriptome of human monocytes involve CTCF. Nucleic Acids Res. 2016, 44, 4090–4104. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Hobaus, J.; Kallay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Engelman, C.D.; Fingerlin, T.E.; Langefeld, C.D.; Hicks, P.J.; Rich, S.S.; Wagenknecht, L.E.; Bowden, D.W.; Norris, J.M. Genetic and environmental determinants of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels in Hispanic and African Americans. J. Clin. Endocrinol. Metab. 2008, 93, 3381–3388. [Google Scholar] [CrossRef] [PubMed]
- Snellman, G.; Melhus, H.; Gedeborg, R.; Olofsson, S.; Wolk, A.; Pedersen, N.L.; Michaelsson, K. Seasonal genetic influence on serum 25-hydroxyvitamin D levels: A twin study. PLoS ONE 2009, 4, e7747. [Google Scholar] [CrossRef]
- Standahl Olsen, K.; Rylander, C.; Brustad, M.; Aksnes, L.; Lund, E. Plasma 25 hydroxyvitamin D level and blood gene expression profiles: A cross-sectional study of the Norwegian Women and Cancer Post-genome Cohort. Eur. J. Clin. Nutr. 2013, 67, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Chavez Valencia, R.A.; Martino, D.J.; Saffery, R.; Ellis, J.A. In vitro exposure of human blood mononuclear cells to active vitamin D does not induce substantial change to DNA methylation on a genome-scale. J Steroid Biochem. Mol. Biol. 2014, 141, 144–149. [Google Scholar] [CrossRef]
- Tapp, H.S.; Commane, D.M.; Bradburn, D.M.; Arasaradnam, R.; Mathers, J.C.; Johnson, I.T.; Belshaw, N.J. Nutritional factors and gender influence age-related DNA methylation in the human rectal mucosa. Aging Cell. 2013, 12, 148–155. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, X.; Shi, H.; Su, S.; Harshfield, G.A.; Gutin, B.; Snieder, H.; Dong, Y. A genome-wide methylation study of severe vitamin D deficiency in African American adolescents. J. Pediatr. 2013, 162, 1004–1009e1. [Google Scholar] [CrossRef]
- Beckett, E.L.; Le Gras, K.; Martin, C.; Boyd, L.; Ng, X.; Duesing, K.; Yates, Z.; Veysey, M.; Lucock, M. Vitamin D Receptor Polymorphisms Relate to Risk of Adenomatous Polyps in a Sex-Specific Manner. Nutr. Cancer. 2016, 68, 193–200. [Google Scholar] [CrossRef]
- Novakovic, B.; Sibson, M.; Ng, H.K.; Manuelpillai, U.; Rakyan, V.; Down, T.; Beck, S.; Fournier, T.; Evain-Brion, D.; Dimitriadis, E.; et al. Placenta-specific methylation of the vitamin D 24-hydroxylase gene: Implications for feedback autoregulation of active vitamin D levels at the fetomaternal interface. In J. Biol. Chem.; 2009; Volume 284, pp. 14838–14848. [Google Scholar]
- Khorchide, M.; Lechner, D.; Cross, H.S. Epigenetic regulation of vitamin D hydroxylase expression and activity in normal and malignant human prostate cells. J. Steroid Biochem. Mol. Biol. 2005, 93, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Armstrong, J.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Dreszer, T.R.; Fujita, P.A.; Guruvadoo, L.; Haeussler, M.; et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015, 43, D670–D681. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, P.; Liel, Y.; Gnainsky, J.; Shany, S.; Schwartz, B. The protective effect of estrogen against chemically induced murine colon carcinogenesis is associated with decreased CpG island methylation and increased mRNA and protein expression of the colonic vitamin D receptor. Oncol. Res. 1999, 11, 255–264. [Google Scholar] [PubMed]
- Lopes, N.; Carvalho, J.; Duraes, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. 2012, 32, 249–257. [Google Scholar] [PubMed]
- Stefanska, B.; Salame, P.; Bednarek, A.; Fabianowska-Majewska, K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br. J. Nutr. 2012, 107, 781–790. [Google Scholar] [CrossRef]
- Rawson, J.B.; Sun, Z.; Dicks, E.; Daftary, D.; Parfrey, P.S.; Green, R.C.; Gallinger, S.; McLaughlin, J.R.; Wang, P.P.; Knight, J.A.; et al. Vitamin D intake is negatively associated with promoter methylation of the Wnt antagonist gene DKK1 in a large group of colorectal cancer patients. Nutr. Cancer. 2012, 64, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeon, S.A.; Kim, B.G.; Takeda, M.; Cho, J.J.; Kim, D.I.; Kawabe, H.; Cho, J.Y. Nedd4 Deficiency in Vascular Smooth Muscle Promotes Vascular Calcification by Stabilizing pSmad1. J. Bone Miner. Res. 2017, 32, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Obeid, R.; Hubner, U.; Bodis, M.; Graeber, S.; Geisel, J. Effect of adding B-vitamins to vitamin D and calcium supplementation on CpG methylation of epigenetic aging markers. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 411–417. [Google Scholar] [CrossRef]
- Glade, M.J. Vitamin D: Health panacea or false prophet? Nutrition 2013, 29, 37–41. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies | Type of Study | Country | Main Findings | Refs |
|---|---|---|---|---|
| Rapson IR, et al. 2017 | Prospective | USA, ARIC study cohort | Deficiency of 25(OH)D was associated with increased risk of PAOD in black and white participants | [23] |
| Liew JY, et al. 2015 | Cross-sectional (case-control) | Australia | No significant difference 25(OH)D levels was detected between PAOD patients (only IC) and control | [32] |
| Veronese N, et al. 2015 | Prospective | Italy, community dwelling men | Baseline hypovitaminosis D (<24 nmol/L) did not predict the onset of PAOD over a 4.4-year follow-up in elderly people | [33] |
| Amer M, et al. 2014 | Retrospective | USA | Elevated serum 25(OH)D concentration was associated with significant increase in ABPI in asymptomatic adults without PAOD | [24] |
| Stricker H, et al. 2012 | Double-blind, placebo-controlled | Caucasian, Switzerland | PAOD patients had low 25(OH)D levels (<30 ng/mL) | [25] |
| McDermott MM, et al. 2012 | Cross-sectional (case-control) | USA | No significant difference 25(OH)D levels was detected between PAOD patients (only IC) and control | [34] |
| Gaddipati VC, et al. 2011 | Cross-sectional | USA | Deficiency of 25(OH)D (<20 ng/mL) was associated with an increased amputation risk in veterans with PAOD | [26] |
| Zagura M, et al. 2011 | Cross-sectional (case-control) | Estonia | PAOD patients had lower 25(OH)D compared to controls | [27] |
| Melamed ML, et al. 2008 | Cross-sectional (case-control) | USA, nationally representative adults >20years of age | Lower serum 25(OH)D levels are associated with a higher prevalence of PAOD | [28] |
| Reis JP, et al. 2008 | Cross-sectional | USA | After adjustment for 25(OH)D levels, odds for PAOD were reduced from 2.11 (95% CI: 1.55, 2.87) to 1.33 (95% CI: 0.84, 2.10) in black compared with white participants | [29] |
| Fahrleitner-Pammer, et al. 2005 | Cross-sectional (case-control) | Austria | Patients with CLI symptoms had lower 25(OH)D compared to both IC and controls | [30] |
| Fahrleitner A, et al. 2002 | Cross-sectional (case-control) | Austria | Patients with CLI symptoms had lower 25(OH)D compared to both IC and controls | [31] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishna, S.M. Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation. Int. J. Mol. Sci. 2019, 20, 4907. https://doi.org/10.3390/ijms20194907
Krishna SM. Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation. International Journal of Molecular Sciences. 2019; 20(19):4907. https://doi.org/10.3390/ijms20194907
Chicago/Turabian StyleKrishna, Smriti Murali. 2019. "Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation" International Journal of Molecular Sciences 20, no. 19: 4907. https://doi.org/10.3390/ijms20194907
APA StyleKrishna, S. M. (2019). Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation. International Journal of Molecular Sciences, 20(19), 4907. https://doi.org/10.3390/ijms20194907