Map-Based Functional Analysis of the GhNLP Genes Reveals Their Roles in Enhancing Tolerance to N-Deficiency in Cotton

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Sequence Analysis of the Cotton NLP Proteins
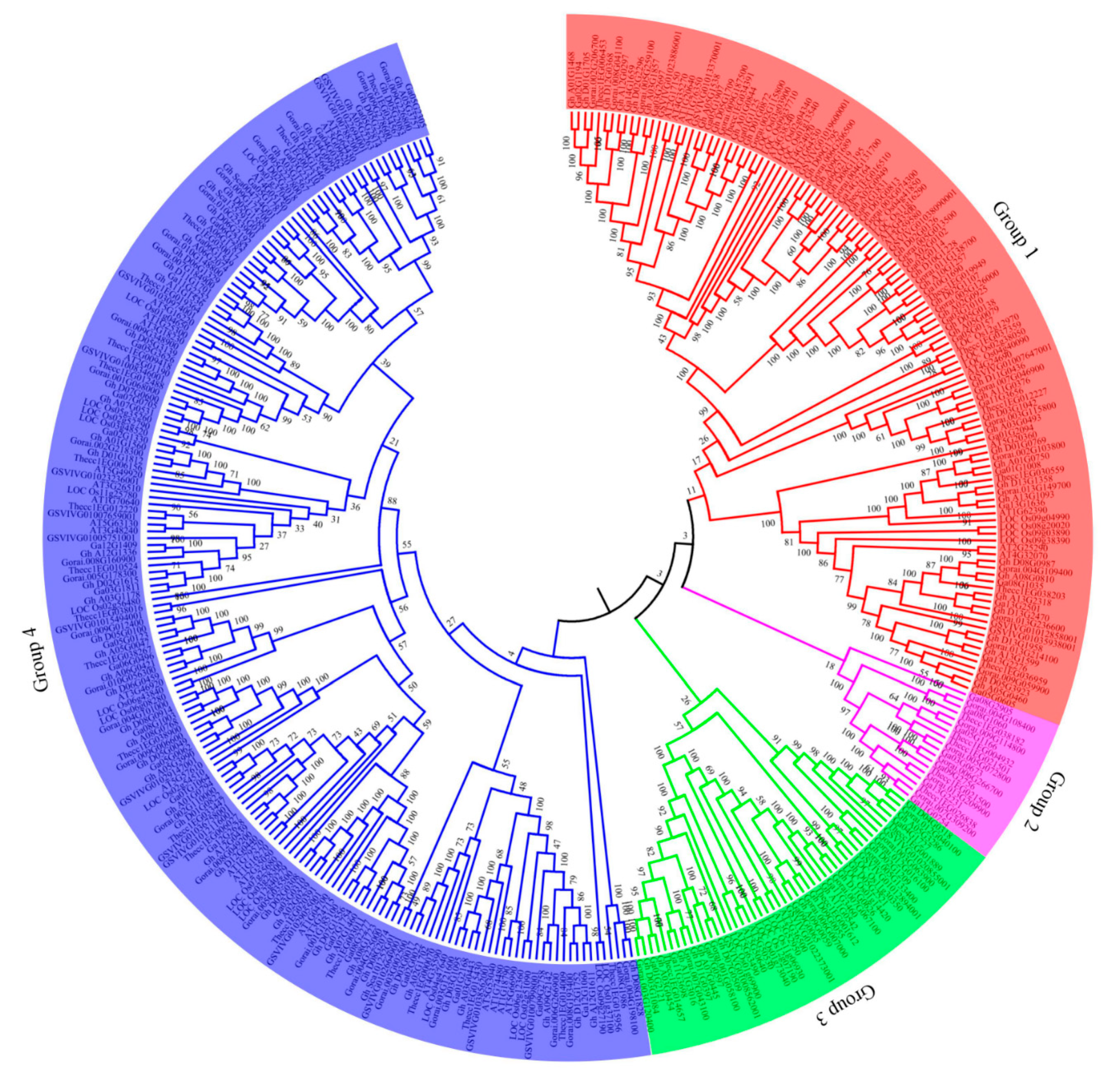
2.2. Phylogenetic Tree Analysis
2.3. Chromosome Mapping and Subcellular Localization Prediction Analysis of the Cotton Proteins Encoded by the NLP Genes in Cotton
2.4. Gene Structure Analysis of the Cotton NLP Proteins
2.5. Cis-regulatory Element Analysis and miRNA Target Prediction on the Cotton NLP Genes
2.6. RT-qPCR Validation of the Selected GhNLP Genes
2.7. Silencing of Gh_A05G3286 (NLP5), Physiological and Morphological Evaluation of the VIGS-Plants and the Non-Cotton Seedlings Under Nitrogen Limited Condition
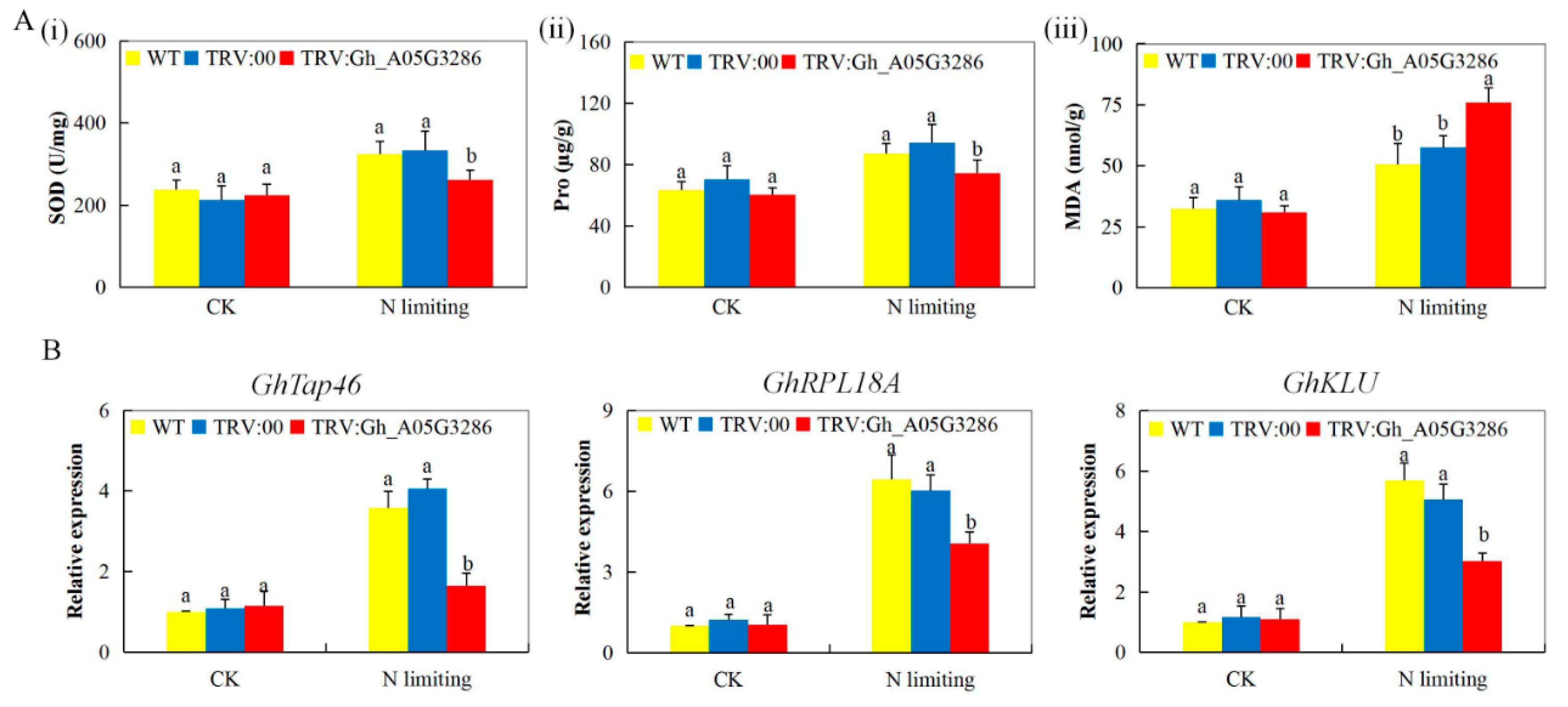
2.8. Transcripts Investigation of Nitrogen Stress-Responsive Genes, analysis of Oxidant, and Antioxidant Content on the Tissues of VIGS and Non-VIGS Cotton Seedling Exposed to Nitrogen Limited Condition
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Identification and Protein Physiochemical Properties Analysis of the NLP Family Genes in Plants
4.3. Phylogenetic Analysis of the NLP Family in Cotton Species with Other Plants
4.4. Chromosomal Locations, Subcellular Localization Prediction, and Gene Structure Analysis
4.5. Cis-regulatory Element Analysis and the miRNA Target Prediction of the Cotton NLP Proteins
4.6. RNA Isolation and Quantitative Reverse-Transcription PCR
4.7. Generation of Transiently Transformed G. hirsutum Plants with Repression of Gh_A05G3286 (NLP5)
4.8. Evaluation of Physiological, Morphological, Biochemical Traits and Expression Analysis of the N-Limited Responsive Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NLP | NODULE-INCEPTION-like proteins |
| VIGS | virus-induced gene silencing |
| GRAVY | Grand hydropathy values |
| SOD | Superoxide dismutase |
| MDA | Malondialdehyde |
| NIN | Nodule inception protein |
References
- Ohyama, T. Nitrogen as a major essential element of plants. Nitrogen Assim. Plants 2010, 37, 1–17. [Google Scholar]
- Qureshi, M.I.; Muneer, S.; Bashir, H.; Ahmad, J.; Iqbal, M. Nodule Physiology and Proteomics of Stressed Legumes. Adv. Bot. Res. 2010, 56, 1–48. [Google Scholar] [CrossRef]
- Kawaharada, Y.; James, E.K.; Kelly, S.; Sandal, N.; Stougaard, J. The Ethylene Responsive Factor Required for Nodulation 1 (ERN1) Transcription Factor Is Required for Infection-Thread Formation in Lotus japonicus. Mol. Plant-Microbe Interact. 2017, 30, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Eyhorn, F.; Ramakrishnan, M.; Mäder, P. The viability of cotton-based organic farming systems in India. Int. J. Agric. Sustain. 2007, 5, 25–38. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, X.; Sun, Y.; Cheng, Y.; Liu, S.; Chen, X.; Feng, G.; Kuyper, T.W. Arbuscular mycorrhizal fungi negatively affect nitrogen acquisition and grain yield of maize in a N deficient soil. Front. Microbiol. 2018, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chang, W.; Fan, Y.; Sun, W.; Qu, C.; Zhang, K.; Liu, L.; Xu, X.; Tang, Z.; Li, J.; et al. Genome-Wide Identification and Characterization of NODULE-INCEPTION-Like Protein (NLP) Family Genes in Brassica napus. Int. J. Mol. Sci. 2018, 19, 2270. [Google Scholar] [CrossRef]
- Olk, D.C. Organic Forms of Soil Nitrogen. Nitrogen Agric. Syst. 2008, 57–100. [Google Scholar] [CrossRef]
- Huppe, H.C.; Turpin, D.H. Integration of Carbon and Nitrogen Metabolism in Plant and Algal Cells. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1994, 45, 577–607. [Google Scholar] [CrossRef]
- Lin, J.S.; Li, X.; Luo, Z.; Mysore, K.S.; Wen, J.; Xie, F. NIN interacts with NLPs to mediate nitrate inhibition of nodulation in Medicago truncatula. Nat. Plants 2018, 4, 942–952. [Google Scholar] [CrossRef]
- Ge, M.; Liu, Y.; Jiang, L.; Wang, Y.; Lv, Y.; Zhou, L.; Liang, S.; Bao, H.; Zhao, H. Genome-wide analysis of maize NLP transcription factor family revealed the roles in nitrogen response. Plant Growth Regul. 2018, 84, 95–105. [Google Scholar] [CrossRef]
- Cao, H.; Qi, S.; Sun, M.; Li, Z.; Yang, Y.; Crawford, N.M.; Wang, Y. Overexpression of the Maize ZmNLP6 and ZmNLP8 Can Complement the Arabidopsis Nitrate Regulatory Mutant nlp7 by Restoring Nitrate Signaling and Assimilation. Front. Plant Sci. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.M.; Ulaganathan, K. Nitrogen Nutrition, Its Regulation and Biotechnological Approaches to Improve Crop Productivity. Am. J. Plant Sci. 2015, 6, 2745–2798. [Google Scholar] [CrossRef] [Green Version]
- Krapp, A.; David, L.C.; Chardin, C.; Girin, T.; Marmagne, A.; Leprince, A.S.; Chaillou, S.; Ferrario-Méry, S.; Meyer, C.; Daniel-Vedele, F. Nitrate transport and signalling in Arabidopsis. J. Exp. Bot. 2014, 65, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xing, X.; Wang, Y.; Tran, A.; Crawford, N.M. A Genetic Screen for Nitrate Regulatory Mutants Captures the Nitrate Transporter Gene NRT1.1. Plant Physiol. 2009, 151, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.C.; Wang, Y.Y.; Tsay, Y.F. AtCIPK8, a CBL-interacting protein kinase, regulates the low-affinity phase of the primary nitrate response. Plant J. 2009, 57, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Mounier, E.; Pervent, M.; Ljung, K.; Gojon, A.; Nacry, P. Auxin-mediated nitrate signalling by NRT1.1 participates in the adaptive response of Arabidopsis root architecture to the spatial heterogeneity of nitrate availability. Plant Cell Environ. 2014, 37, 162–174. [Google Scholar] [CrossRef]
- Liu, X.; Huang, D.; Tao, J.; Miller, A.J.; Fan, X.; Xu, G. Identification and functional assay of the interaction motifs in the partner protein OsNAR2.1 of the two-component system for high-affinity nitrate transport. New Phytol. 2014, 204, 74–80. [Google Scholar] [CrossRef]
- Gu, R.; Duan, F.; An, X.; Zhang, F.; Von Wirén, N.; Yuan, L. Characterization of AMT-mediated high-affinity ammonium uptake in roots of maize (Zea mays L.). Plant Cell Physiol. 2013, 54, 1515–1524. [Google Scholar] [CrossRef]
- Okamoto, M.; Vidmar, J.J.; Glass, A.D.M. Regulation of NRT1 and NRT2 gene families of Arabidopsis thaliana: Responses to nitrate provision. Plant Cell Physiol. 2003, 44, 304–317. [Google Scholar] [CrossRef]
- Lezhneva, L.; Kiba, T.; Feria-Bourrellier, A.B.; Lafouge, F.; Boutet-Mercey, S.; Zoufan, P.; Sakakibara, H.; Daniel-Vedele, F.; Krapp, A. The Arabidopsis nitrate transporter NRT2.5 plays a role in nitrate acquisition and remobilization in nitrogen-starved plants. Plant J. 2014, 80, 230–241. [Google Scholar] [CrossRef]
- Mueller, N.D.; Gerber, J.S.; Johnston, M.; Ray, D.K.; Ramankutty, N.; Foley, J.A. Closing yield gaps through nutrient and water management. Nature 2012, 490, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Garnett, T.; Conn, V.; Kaiser, B.N. Root based approaches to improving nitrogen use efficiency in plants. Plant Cell Environ. 2009, 32, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Bharali, B.; Haloi, B.; Chutia, J.; Chack, S.; Hazarika, K. Susceptibility of Some Wheat (Triticum aestivum L.) Varieties to Aerosols of Oxidised and Reduced Nitrogen. Adv. Crop Sci. Technol. 2015, 3, 4. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Goulao, L.F.; Amâncio, S. Regulation of cell wall remodeling in grapevine (Vitis vinifera L.) callus under individual mineral stress deficiency. J. Plant Physiol. 2016, 190, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.T.; Rehman, A. Overview on Effects of Water Stress on Cotton Plants and Productivity. In Biochemical, Physiological and Molecular Avenues for Combating Abiotic Stress Tolerance in Plants; Academic Press: London, UK, 2018; pp. 297–316. [Google Scholar]
- Paterson, A.H.; May, O.L.; Desai, A.; Rong, J.; Chee, P.W. Chromosome structural changes in diploid and tetraploid A genomes of Gossypium. Genome 2006, 49, 336–345. [Google Scholar] [CrossRef]
- Meshram, L.D.; Tayyab, M.A. Cytomorphology of two interspecific hybrids of Gossypium. PKV Res. J. 1994, 18, 63–67. [Google Scholar]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Schauser, L.; Wieloch, W.; Stougaard, J. Evolution of NIN-like proteins in Arabidopsis, rice, and Lotus japonicus. J. Mol. Evol. 2005, 60, 229–237. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, F.; Chen, D.; Chu, W.; Liu, H.; Xiang, Y. Genome-wide identification and analysis of the Populus trichocarpa TIFY gene family. Plant Physiol. Biochem. 2017, 115, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.; Wang, X.; Cai, X.; Zhou, Z.; Zhang, Z.; Salih, H.; Wang, K.; et al. Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, R.; Pareek, A.; Sopory, S.K.; Singla-Pareek, S.L. Overexpression of rice CBS domain containing protein improves salinity, oxidative, and heavy metal tolerance in transgenic tobacco. Mol. Biotechnol. 2012, 52, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.N.; Shang, W.; Zhang, C.; Chen, H.; Chen, L.; Yuan, S.; Chen, S.; Zhang, X.; Zhou, X. Identification and comparative analysis of CBS domain-containing proteins in soybean (Glycine max) and the primary function of GmCBS21 in enhanced tolerance to low nitrogen stress. Int. J. Mol. Sci. 2016, 17, 620. [Google Scholar] [CrossRef] [PubMed]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Kirungu, J.N.; Lu, P.; Yang, X.; Dong, Q.; Cai, X.; Xu, Y.; Wang, X.; Zhou, Z.; Hou, Y.; et al. Genome wide identification of the trihelix transcription factors and overexpression of Gh_A05G2067 (GT-2), a novel gene contributing to increased drought and salt stresses tolerance in cotton. Physiol. Plant. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Yogindran, S.; Rajam, M.V. Role of miRNAs in biotic stress reactions in plants. Indian J. Plant Physiol. 2017, 22, 514–529. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, J.; Zhu, Q.; Zhao, L.; Sui, S.; Li, Z.; Zhang, Y.; Wang, H.; Tian, D.; Zhao, Y. Identification of microRNAs involved in drought stress responses in early-maturing cotton by high-throughput sequencing. Genes Genom. 2018, 40, 305–314. [Google Scholar] [CrossRef]
- Li, J.; Guo, G.; Guo, W.; Guo, G.; Tong, D.; Ni, Z.; Sun, Q.; Yao, Y. miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea mays L.). BMC Plant Biol. 2012, 12, 220. [Google Scholar] [CrossRef]
- Zhao, M.; Ding, H.; Zhu, J.K.; Zhang, F.; Li, W.X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol. 2011, 190, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Scofield, S.R.; Huang, L.; Brandt, A.S.; Gill, B.S. Development of a Virus-Induced Gene-Silencing System for Hexaploid Wheat and Its Use in Functional Analysis of the Lr21-Mediated Leaf Rust Resistance Pathway. Plant Physiol. 2005, 138, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Estévez, J.M.; Cantero, A.; Romero, C.; Kawaide, H.; Jiménez, L.F.; Kuzuyama, T.; Seto, H.; Kamiya, Y.; León, P. Analysis of the Expression of CLA1, a Gene That Encodes the 1-Deoxyxylulose 5-Phosphate Synthase of the 2-C-Methyl-d-Erythritol-4-Phosphate Pathway in Arabidopsis. Plant Physiol. 2002, 124, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.V.; Andrade-Navarro, M.A.; Ketting, R.F. Function and Evolution of Nematode RNAi Pathways. Non-Coding RNA 2019, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Dong, Q.; Cai, X.; Zhou, Z.; Wang, X.; Hou, Y.; Xu, Y.; Peng, R.; et al. Knockdown of Cytochrome P450 Genes Gh_D07G1197 and Gh_A13G2057 on Chromosomes D07 and A13 Reveals Their Putative Role in Enhancing Drought and Salt Stress Tolerance in Gossypium hirsutum. Genes 2019, 10, 226. [Google Scholar] [CrossRef]
- Bachan, S.; Dinesh-Kumar, S.P. Tobacco rattle virus (TRV)-based virus-induced gene silencing. Methods Mol. Biol. 2012, 894, 83–92. [Google Scholar] [CrossRef]
- Ding, L.; Wang, K.J.; Jiang, G.M.; Biswas, D.K.; Xu, H.; Li, L.F.; Li, Y.H. Effects of nitrogen deficiency on photosynthetic traits of maize hybrids released in different years. Ann. Bot. 2005, 96, 925–930. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Mohapatra, P.K.; Fujita, K.; Nakabayashi, K.; Thompson, J. Effect of nitrogen deficiency on biomass production, photosynthesis, carbon partitioning, and nitrogen nutrition status of Melaleuca and Eucalyptus species. Soil Sci. Plant Nutr. 2003, 49, 99–109. [Google Scholar] [CrossRef]
- Løvdal, T.; Lillo, C. Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal. Biochem. 2009, 387, 238–242. [Google Scholar] [CrossRef]
- Warzybok, A.; Migocka, M. Reliable Reference Genes for Normalization of Gene Expression in Cucumber Grown under Different Nitrogen Nutrition. PLoS ONE 2013, 8, e72887. [Google Scholar] [CrossRef]
- Fuentes, S.I.; Allen, D.J.; Ortiz-Lopez, A.; Hernández, G. Over-expression of cytosolic glutamine synthetase increases photosynthesis and growth at low nitrogen concentrations. J. Exp. Bot. 2001, 52, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Lee, J.; Kichey, T.; Gerentes, D.; Zivy, M.; Tatout, C.; Dubois, F.; Balliau, T.; Valot, B.; Davanture, M.; et al. Two Cytosolic Glutamine Synthetase Isoforms of Maize Are Specifically Involved in the Control of Grain Production. Plant Cell 2006, 18, 3252–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liheng, H.; Zhiqiang, G.; Runzhi, L. Pretreatment of seed with H2O2 enhances drought tolerance of wheat (Triticum aestivum L.) seedlings. Afr. J. Biotechnol. 2009, 8, 6151–6157. [Google Scholar] [CrossRef]
- Smirnoff, N.; Arnaud, D. Hydrogen peroxide metabolism and functions in plants. New Phytol. 2019, 221, 1197–1214. [Google Scholar] [CrossRef]
- Matsuki, S.; Ogawa, K.; Tanaka, A.; Hara, T. Morphological and photosynthetic responses of Quercus crispula seedlings to high-light conditions. Tree Physiol. 2003, 23, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Losciale, P.; Zibordi, M.; Manfrini, L.; Morandi, B.; Corelli-Grappadelli, L. Effect of moderate light reduction on absorbed energy management, water use, photoprotection and photo-damage in peach. Acta Hortic. 2011, 907, 169–174. [Google Scholar] [CrossRef]
- Yevenes, M.A.; Mannaerts, C.M. Seasonal and land use impacts on the nitrate budget and export of a mesoscale catchment in Southern Portugal. Agric. Water Manag. 2011, 102, 54–65. [Google Scholar] [CrossRef]
- Bushong, J.T.; Arnall, D.B.; Raun, W.R. Effect of Preplant Irrigation, Nitrogen Fertilizer Application Timing, and Phosphorus and Potassium Fertilization on Winter Wheat Grain Yield and Water Use Efficiency. Int. J. Agron. 2014, 2014. [Google Scholar] [CrossRef]
- Chao, Z.; Zhang, P. Assessing the impact of urbanization on vegetation change and arable land resources change in Shandong province. In Proceedings of the 3rd International Conference on Agro-Geoinformatics, Beijing, China, 11–14 August 2014. [Google Scholar]
- Suzuki, W.; Konishi, M.; Yanagisawa, S. The evolutionary events necessary for the emergence of symbiotic nitrogen fixation in legumes may involve a loss of nitrate responsiveness of the NIN transcription factor. Plant Signal. Behav. 2013, 8, e25975. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Zhang, G.; Sun, L.; Wang, J.; Hao, F. Genome-wide identification of CBL family and expression analysis of CBLs in response to potassium deficiency in cotton. PeerJ 2017, 5, e3653. [Google Scholar] [CrossRef]
- Hachiya, T.; Mizokami, Y.; Miyata, K.; Tholen, D.; Watanabe, C.K.; Noguchi, K. Evidence for a nitrate-independent function of the nitrate sensor NRT1.1 in Arabidopsis thaliana. J. Plant Res. 2011, 124, 425–430. [Google Scholar] [CrossRef]
- Bush, M.; Dixon, R. The Role of Bacterial Enhancer Binding Proteins as Specialized Activators of 54-Dependent Transcription. Microbiol. Mol. Biol. Rev. 2012, 76, 497–529. [Google Scholar] [CrossRef] [PubMed]
- Chardin, C.; Girin, T.; Roudier, F.; Meyer, C.; Krapp, A. The plant RWP-RK transcription factors: Key regulators of nitrogen responses and of gametophyte development. J. Exp. Bot. 2014, 65, 5577–5587. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Magwanga, R.O.; Guo, X.; Kirungu, J.N.; Lu, H.; Cai, X.; Zhou, Z.; Wei, Y.; Wang, X.; Zhang, Z.; et al. Genome-Wide Analysis of Multidrug and Toxic Compound Extrusion (MATE) Family in Diploid Cotton, Gossypium raimondii and Gossypium arboreum and Its Expression Analysis Under Salt, Cadmium and Drought Stress. G3 2018, 8, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Morselli, M.; Pastor, W.A.; Montanini, B.; Nee, K.; Ferrari, R.; Fu, K.; Bonora, G.; Rubbi, L.; Clark, A.T.; Ottonello, S.; et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife 2015, 4, e06205. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Taylor, D.R. Testing for selection on synonymous sites in plant mitochondrial DNA: The role of codon bias and RNA editing. J. Mol. Evol. 2010, 70, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Rauwolf, U.; Meurer, J.; Herrmann, R.G. The role of plastids in plant speciation. Mol. Ecol. 2011, 20, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Ondřej, V.; Lukášová, E.; Krejčí, J.; Kozubek, S. Intranuclear trafficking of plasmid DNA is mediated by nuclear polymeric proteins lamins and actin. Acta Biochim. Pol. 2008, 55, 307–315. [Google Scholar] [PubMed]
- Stanbrough, M.; Rowen, D.W.; Magasanik, B. Role of the GATA factors Gln3p and Nil1p of Saccharomyces cerevisiae in the expression of nitrogen-regulated genes. Proc. Natl. Acad. Sci. USA 1995, 92, 9450–9454. [Google Scholar] [CrossRef]
- Beck, T.; Hall, M.N. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 1999, 402, 689–692. [Google Scholar] [CrossRef]
- Chen, M.; Bao, H.; Wu, Q.; Wang, Y. Transcriptome-wide identification of miRNA targets under nitrogen deficiency in Populus tomentosa using degradome sequencing. Int. J. Mol. Sci. 2015, 16, 13937–13958. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Hao, Q.; Sha, A.; Zhou, R.; Zhou, X.; Yuan, L. Elucidation of miRNAs-Mediated Responses to Low Nitrogen Stress by Deep Sequencing of Two Soybean Genotypes. PLoS ONE 2013, 8, e67423. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; He, H.; Yu, D. Identification of Nitrogen Starvation-Responsive MicroRNAs in Arabidopsis thaliana. PLoS ONE 2012, 7, e48951. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Z.; Mo, Q.; Zou, C.; Li, W.; Xu, Y.; Xie, C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot. 2013, 112, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, H.; Hamera, S.; Chen, X.; Fang, R. MiR444a has multiple functions in the rice nitrate-signaling pathway. Plant J. 2014, 78, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-F.; Tian, Q.; Reed, J.W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef]
- Gifford, M.L.; Dean, A.; Gutierrez, R.A.; Coruzzi, G.M.; Birnbaum, K.D. Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad. Sci. USA 2008, 105, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef]
- Guo, H.-S. MicroRNA Directs mRNA Cleavage of the Transcription Factor NAC1 to Downregulate Auxin Signals for Arabidopsis Lateral Root Development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef]
- Mishra, A.K.; Duraisamy, G.S.; Matoušek, J. Discovering MicroRNAs and Their Targets in Plants. CRC Crit. Rev. Plant Sci. 2015, 34, 553–571. [Google Scholar] [CrossRef]
- Xu, Z.; Yu, J.Z.; Cho, J.; Yu, J.; Kohel, R.J.; Percy, R.G. Polyploidization Altered Gene Functions in Cotton (Gossypium spp.). PLoS ONE 2010, 5, e14351. [Google Scholar] [CrossRef] [PubMed]
- Abdelraheem, A.; Fang, D.D.; Zhang, J. Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of Upland cotton under the greenhouse and field conditions. Euphytica 2018, 214, 8. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.J.; Thaxton, P.M.; El-Zik, K.M.; Paterson, A.H. D-subgenome bias of Xcm resistance genes in tetraploid Gossypium (cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 1998, 149, 1987–1996. [Google Scholar] [PubMed]
- Guo, W.; Cai, C.; Wang, C.; Zhao, L.; Wang, L.; Zhang, T. A preliminary analysis of genome structure and composition in Gossypium hirsutum. BMC Genom. 2008, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Key, J. MAC Significance of mating systems for chromosomes and gametes in polyploids. Hereditas 1970, 66, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.J.; Dong, J.M.; Brubaker, C.L.; Stelly, D.M.; Wendel, J.F.; Paterson, A.H. A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: Chromosome organization and evolution in a disomic polyploid genome. Genetics 1994, 138, 829–847. [Google Scholar]
- Basu, S.; Roychoudhury, A. Expression profiling of abiotic stress-inducible genes in response to multiple stresses in rice (Oryza sativa L.) varieties with contrasting level of stress tolerance. BioMed Res. Int. 2014, 2014, 706890. [Google Scholar] [CrossRef]
- Swindell, W.R. The association among gene expression responses to nine abiotic stress treatments in Arabidopsis thaliana. Genetics 2006, 174, 1811–1824. [Google Scholar] [CrossRef]
- Lin, Y.L.; Chao, Y.Y.; Huang, W.D.; Kao, C.H. Effect of nitrogen deficiency on antioxidant status and Cd toxicity in rice seedlings. Plant Growth Regul. 2011, 64, 263–273. [Google Scholar] [CrossRef]
- Lin, W.; Hagen, E.; Fulcher, A.; Hren, M.T.; Cheng, Z.M. Overexpressing the ZmDof1 gene in Populus does not improve growth and nitrogen assimilation under low-nitrogen conditions. Plant Cell Tissue Organ Cult. 2013, 113, 51–61. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Prsa, I.; Stampar, F.; Vodnik, D.; Veberic, R. Influence of nitrogen on leaf chlorophyll content and photosynthesis of “Golden Delicious” apple. Acta Agric. Scand. Sect. B Soil Plant Sci. 2007, 57, 283–289. [Google Scholar] [CrossRef]
- Ahn, C.S.; Ahn, H.K.; Pai, H.S. Overexpression of the PP2A regulatory subunit Tap46 leads to enhanced plant growth through stimulation of the TOR signalling pathway. J. Exp. Bot. 2015, 66, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.M. Effect of nutrient solution pH and N-Sources (NH4/NO3) on the growth and elemental content of rice plants. Agronomie 1984, 4, 361–365. [Google Scholar] [CrossRef]
- Li, X.Q.; Sveshnikov, D.; Zebarth, B.J.; Tai, H.; de Koeyer, D.; Millard, P.; Haroon, M.; Singh, M. Detection of nitrogen sufficiency in potato plants using gene expression markers. Am. J. Potato Res. 2010, 87, 50–59. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef]
- Gálvez, J.H.; Tai, H.H.; Lagüe, M.; Zebarth, B.J.; Strömvik, M.V. The nitrogen responsive transcriptome in potato (Solanum tuberosum L.) reveals significant gene regulatory motifs. Sci. Rep. 2016, 6, 26090. [Google Scholar] [CrossRef]
- do, A.C.; Bisognin, D.; Steffens, C. Non-destructive quantification of chlorophylls in leaves by means of a colorimetric method. Hortic. Bras. 2008, 26, 471–475. [Google Scholar] [CrossRef]
- Giannopolitis, C.N.; Ries, S.K. Superoxide Dismutases: II. Purification and Quantitative Relationship with Water-soluble Protein in Seedlings. Plant Physiol. 1977, 59, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, F.; Cardinaels, C.; Clijsters, H. Induction of enzyme capacity in plants as a result of heavy metal toxicity: Dose-response relations in Phaseolus vulgaris L., treated with zinc and cadmium. Environ. Pollut. 1988, 52, 103–115. [Google Scholar] [CrossRef]
- Cakmak, I.; Horst, W.J. Effect of aluminium on lipid peroxidation, siiperoxide dismutase, catalase, and peroxidase activities in root tips of soybean (Glycine max). Physiol. Plant. 1991, 83, 463–468. [Google Scholar] [CrossRef]
- Chen, X.; Kou, M.; Tang, Z.; Zhang, A.; Li, H. The use of humic acid urea fertilizer for increasing yield and utilization of nitrogen in sweet potato. Plant Soil Environ. 2017, 63, 201–206. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magwanga, R.O.; Kirungu, J.N.; Lu, P.; Cai, X.; Zhou, Z.; Xu, Y.; Hou, Y.; Agong, S.G.; Wang, K.; Liu, F. Map-Based Functional Analysis of the GhNLP Genes Reveals Their Roles in Enhancing Tolerance to N-Deficiency in Cotton. Int. J. Mol. Sci. 2019, 20, 4953. https://doi.org/10.3390/ijms20194953
Magwanga RO, Kirungu JN, Lu P, Cai X, Zhou Z, Xu Y, Hou Y, Agong SG, Wang K, Liu F. Map-Based Functional Analysis of the GhNLP Genes Reveals Their Roles in Enhancing Tolerance to N-Deficiency in Cotton. International Journal of Molecular Sciences. 2019; 20(19):4953. https://doi.org/10.3390/ijms20194953
Chicago/Turabian StyleMagwanga, Richard Odongo, Joy Nyangasi Kirungu, Pu Lu, Xiaoyan Cai, Zhongli Zhou, Yanchao Xu, Yuqing Hou, Stephen Gaya Agong, Kunbo Wang, and Fang Liu. 2019. "Map-Based Functional Analysis of the GhNLP Genes Reveals Their Roles in Enhancing Tolerance to N-Deficiency in Cotton" International Journal of Molecular Sciences 20, no. 19: 4953. https://doi.org/10.3390/ijms20194953
APA StyleMagwanga, R. O., Kirungu, J. N., Lu, P., Cai, X., Zhou, Z., Xu, Y., Hou, Y., Agong, S. G., Wang, K., & Liu, F. (2019). Map-Based Functional Analysis of the GhNLP Genes Reveals Their Roles in Enhancing Tolerance to N-Deficiency in Cotton. International Journal of Molecular Sciences, 20(19), 4953. https://doi.org/10.3390/ijms20194953