Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
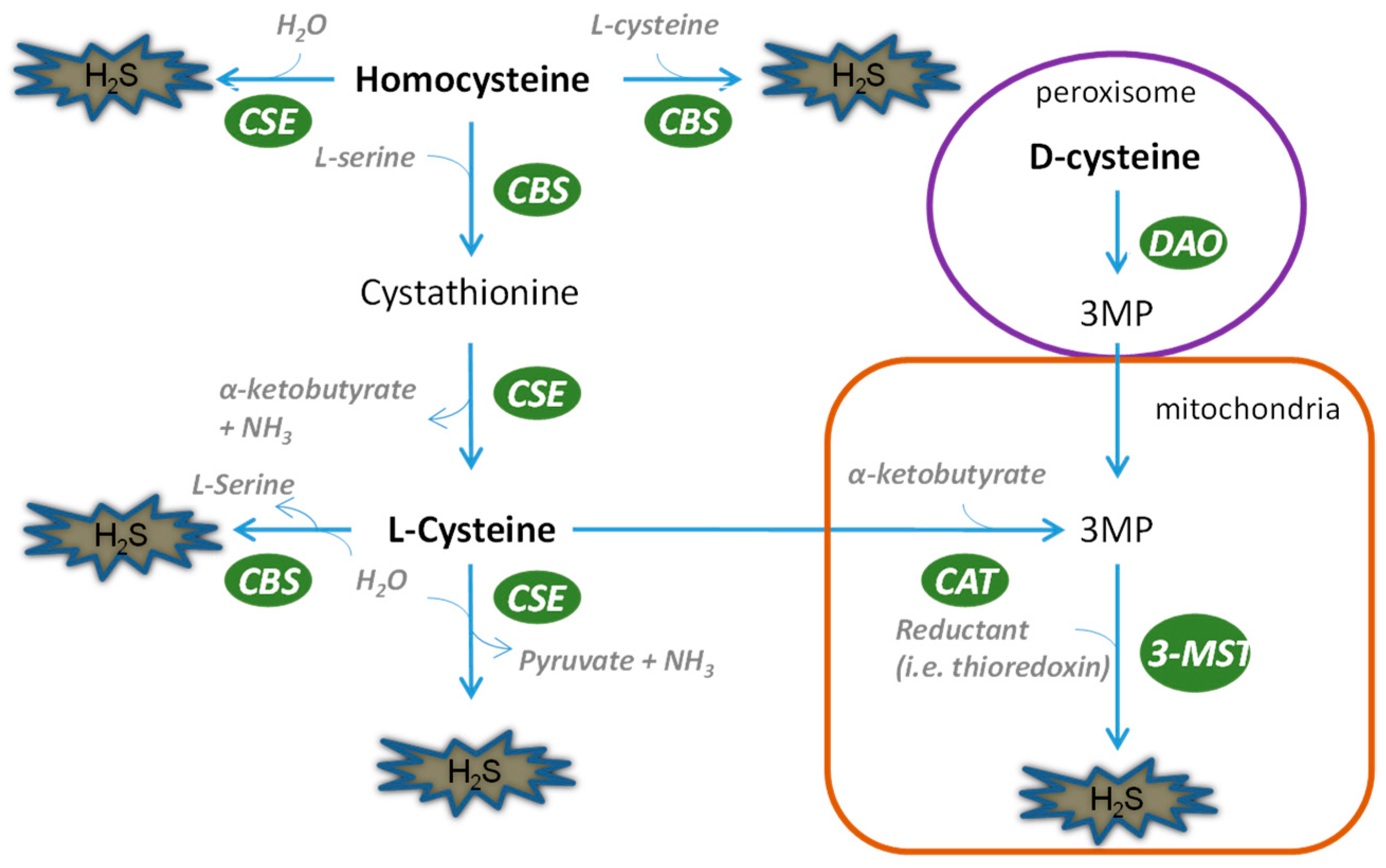
2. Biosynthesis of H2S and Hydrogen Polysulfides in the Kidney
2.1. Biosynthesis of H2S in the Kidney
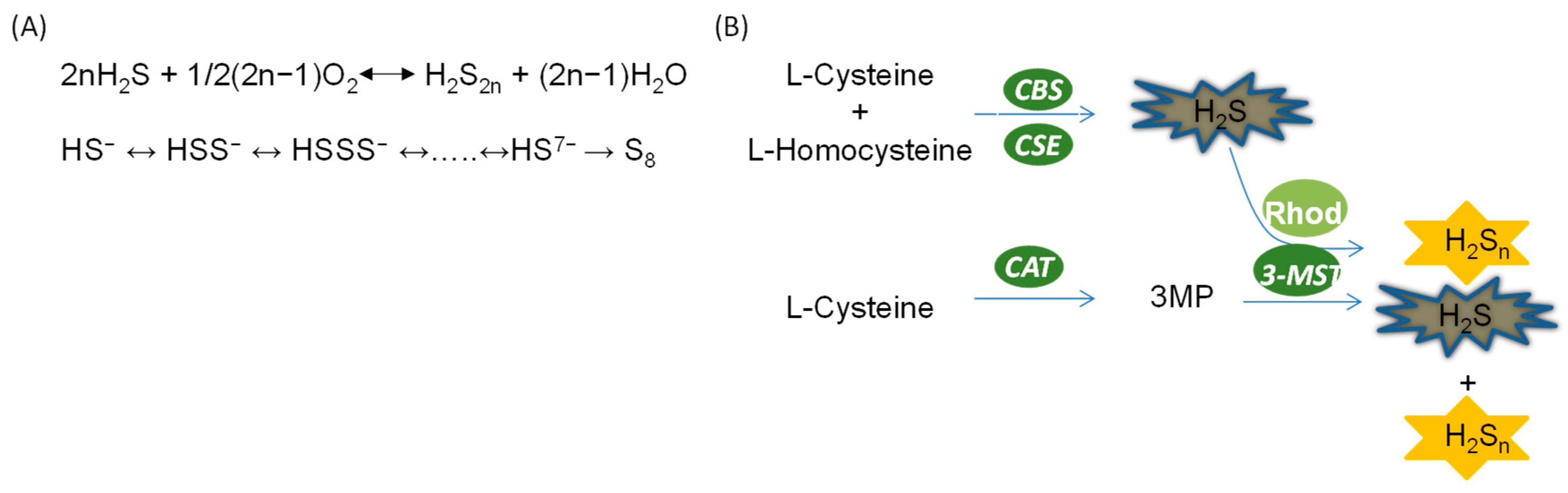
2.2. Biosynthesis of Hydrogen Polysulfide in the Kidney
2.2.1. Non-Enzymatic Generation of Polysulfide
2.2.2. Enzymatic Generation of Polysulfide
3. Cisplatin-Induced Nephrotoxicity
3.1. Clinical Features of Cisplatin-Induced Nephrotoxicity
3.2. Risk Factors of Cisplatin-Induced Nephrotoxicity
3.3. Disease Pathophysiology of Cisplatin-Induced Nephrotoxicity
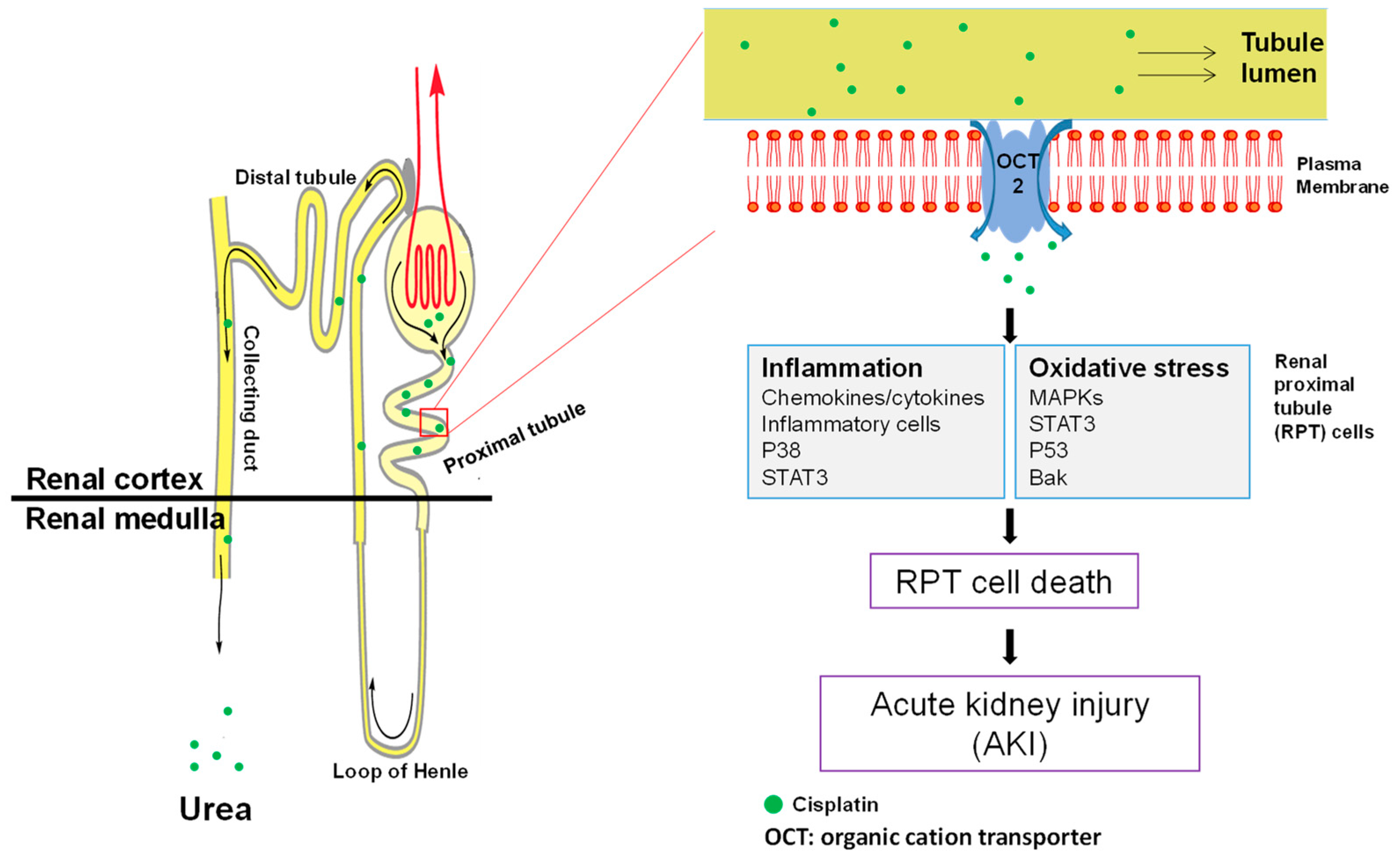
3.3.1. Accumulation of Cisplatin in Kidney Cells
3.3.2. Cell Death in Cisplatin-Induced Nephrotoxicity: Types and Location
3.3.3. Oxidative Stress in Cisplatin-Induced Nephrotoxicity
3.3.4. MAPK Activation in Cisplatin-Induced Nephrotoxicity
3.3.5. Inflammation in Cisplatin-Induced Nephrotoxicity
3.4. Prevention of Cisplatin-Induced Nephrotoxicity
4. Protective Effect of Hydrogen Sulfide in Cisplatin-Induced Nephrotoxicity
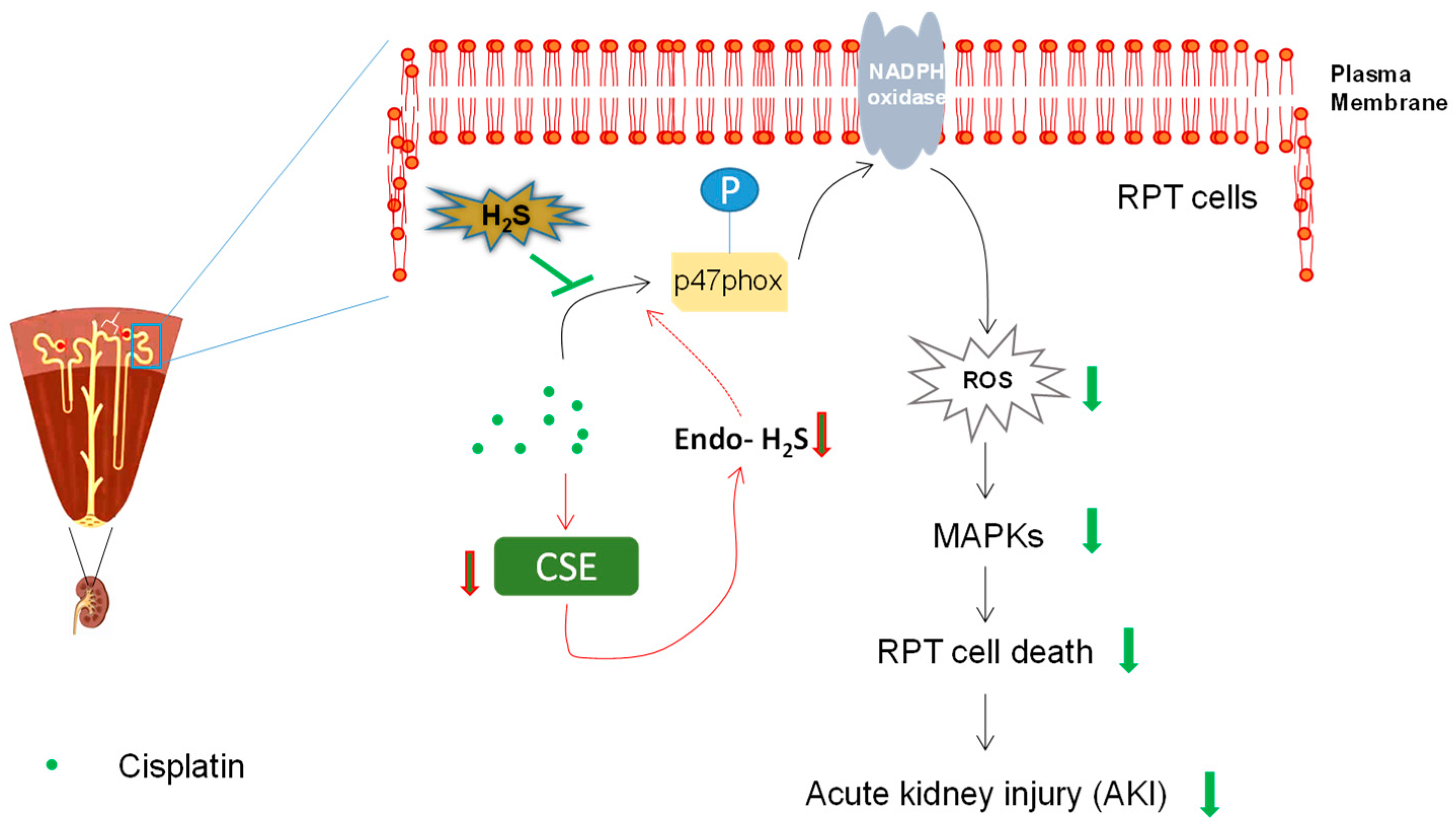
4.1. Role of Endogenous H2S in Cisplatin-Induced Nephrotoxicity
4.2. Donation of H2S Protects Against Cisplatin-Induced Nephrotoxicity
4.3. H2S Exhibited Anti-Oxidant Effect
4.4. H2S Exhibited Anti-Apoptotic Effect
4.5. Can H2S Enhance the Anti-Cancer Effect of Cisplatin?
5. Therapeutic Potential of Polysulfide in Cisplatin-Induced Nephrotoxicity
6. H2S and Polysulfide as A Remedy for Cisplatin-Mediated Toxicity in Other Organs?
7. Future Perspectives and Conclusions
Funding
Conflicts of Interest
Abbreviations
| H2S | Hydrogen sulfide |
| NO | Nitric oxide |
| CO | Carbon monoxide |
| CSE | Cystathionine γ-lyase |
| CBS | Cystathionine β-synthase |
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| CAT | Cysteine aminotransferase |
| DAO | d-amino acid oxidase |
| OCT2 | Organic cation transporter 2 |
| Ctrl1 | Copper transporter 1 |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| NAC | N-Acetylcysteine |
| RPT | Renal proximal tubule |
| DADS | Diallyl disulfides |
| DATS | Diallyl trisulfides |
References
- Smith, R.P.; Gosselin, R.E. Hydrogen sulfide poisoning. J. Occup. Med. Off. Publ. Ind. Med. Assoc. 1979, 21, 93–97. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’sa crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhang, Y.; Yang, M.; Wang, S.; Jiang, Z.; Li, Z. Exogenous hydrogen sulfide prevents kidney damage following unilateral ureteral obstruction. Neurourol. Urodyn. 2014, 33, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Ren. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef] [PubMed]
- Ahangarpour, A.; Abdollahzade Fard, A.; Gharibnaseri, M.K.; Jalali, T.; Rashidi, I. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. Vet. Res. Forum Int. Q. J. 2014, 5, 121–127. [Google Scholar]
- Karimi, A.; Absalan, F.; Khorsandi, L.; Valizadeh, A.; Mansouri, E. Sodium hydrogen sulfide (NaHS) ameliorates alterations caused by cisplatin in filtration slit diaphragm and podocyte cytoskeletal in rat kidney. J. Nephropathol. 2017, 6, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Bian, J.S. The Role of Hydrogen Sulfide in Renal System. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef]
- Dugbartey, G.J. The smell of renal protection against chronic kidney disease: Hydrogen sulfide offers a potential stinky remedy. Pharmacol. Rep. 2018, 70, 196–205. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Feliers, D.; Lee, H.J. Hydrogen sulfide as a regulatory factor in kidney health and disease. Biochem. Pharmacol. 2018, 149, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Winterbourn, C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Toyofuku, Y.; Koike, S.; Shibuya, N.; Nagahara, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci. Rep. 2015, 5, 14774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Ries, F.; Klastersky, J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am. J. Kidney Dis. 1986, 8, 368–379. [Google Scholar] [CrossRef]
- Hill, J.; Speer, R. Organo-platinum complexes as antitumor agents (review). Anticancer Res. 1981, 2, 173–186. [Google Scholar]
- Kociba, R.J.; Sleight, S. Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemother. Rep. Part 1 1971, 55, 1–8. [Google Scholar]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- Santoso, J.T.; Lucci, J.A., III; Coleman, R.L.; Schafer, I.; Hannigan, E.V. Saline, mannitol, and furosemide hydration in acute cisplatin nephrotoxicity: A randomized trial. Cancer Chemother. Pharmacol. 2003, 52, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Nazneen, A.; Abid, M.; Razzaque, M. Cisplatin-associated nephrotoxicity and pathological events. In Cellular Stress Responses in Renal Diseases; Karger Publishers: Basel, Switzerland, 2005; Volume 148, pp. 107–121. [Google Scholar]
- Reece, P.A.; Stafford, I.; Russell, J.; Khan, M.; Gill, P. Creatinine clearance as a predictor of ultrafilterable platinum disposition in cancer patients treated with cisplatin: Relationship between peak ultrafilterable platinum plasma levels and nephrotoxicity. J. Clin. Oncol. 1987, 5, 304–309. [Google Scholar] [CrossRef]
- Madias, N.E.; Harrington, J.T. Platinum nephrotoxicity. Am. J. Med. 1978, 65, 307–314. [Google Scholar] [CrossRef]
- De Jongh, F.E.; Verweij, J.; Loos, W.J.; de Wit, R.; de Jonge, M.J.; Planting, A.S.; Nooter, K.; Stoter, G.; Sparreboom, A. Body-surface area–based dosing does not increase accuracy of predicting cisplatin exposure. J. Clin. Oncol. 2001, 19, 3733–3739. [Google Scholar] [CrossRef]
- De Jongh, F.; Van Veen, R.; Veltman, S.; de Wit, R.; Van der Burg, M.; Van den Bent, M.; Planting, A.; Graveland, W.; Stoter, G.; Verweij, J. Weekly high-dose cisplatin is a feasible treatment option: Analysis on prognostic factors for toxicity in 400 patients. Br. J. Cancer 2003, 88, 1199–1206. [Google Scholar] [CrossRef]
- Scott, L.A.; Madan, E.; Valentovic, M.A. Attenuation of cisplatin nephrotoxicity by streptozotocin-induced diabetes. Fundam. Appl. Toxicol. 1989, 12, 530–539. [Google Scholar] [CrossRef]
- Gogas, H.; Shapiro, F.; Aghajanian, C.; Fennelly, D.; Almadrones, L.; Hoskins, W.; Spriggs, D. The impact of diabetes mellitus on the toxicity of therapy for advanced ovarian cancer. Gynecol. Oncol. 1996, 61, 22–26. [Google Scholar] [CrossRef]
- Stewart, D.J.; Dulberg, C.S.; Mikhael, N.Z.; Redmond, M.D.; Montpetit, V.A.; Goel, R. Association of cisplatin nephrotoxicity with patient characteristics and cisplatin administration methods. Cancer Chemother. Pharmacol. 1997, 40, 293–308. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of Organic Cation Transporter 2 (OCT2) to Cisplatin-Induced Nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H. Organic cation transporter 2 mediates cisplatin-induced oto-and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Safirstein, R.; Miller, P.; Guttenplan, J.B. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984, 25, 753–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, R.J.; Ghazi, M.A.; Barfuss, D.W. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl-l-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother. Pharmacol. 2003, 51, 132–138. [Google Scholar] [PubMed]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol.-Ren. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, T.; Riethmüller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. American journal of physiology. Ren. Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef]
- Lieberthal, W.; Triaca, V.; Levine, J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: Apoptosis vs. necrosis. Am. J. Physiol. 1996, 270, F700–F708. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baliga, R.; Ueda, N.; Walker, P.D.; Shah, S.V. Oxidant mechanisms in toxic acute renal failure. Drug Metab. Rev. 1999, 31, 971–997. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Safirstein, R.L. Cisplatin Nephrotoxicity, Seminars in Nephrology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 460–464. [Google Scholar]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Rashed, L.A.; Hashem, R.M.; Soliman, H.M. Oxytocin inhibits NADPH oxidase and P38 MAPK in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 2011, 65, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.; Molina-Jijon, E.; Medina-Campos, O.N.; Rodriguez-Munoz, R.; Reyes, J.L.; Barrera, D.; Pedraza-Chaverri, J. Superoxide anion production and expression of gp91(phox) and p47(phox) are increased in glomeruli and proximal tubules of cisplatin-treated rats. J. Biochem. Mol. Toxicol. 2015, 29, 149–156. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Pan, H.; Huang, W.; Wang, X.; Wen, H.; Shen, K.; Jin, B. Pharmacological inhibition of NADPH oxidase protects against cisplatin induced nephrotoxicity in mice by two step mechanism. Food Chem. Toxicol. 2015, 83, 251–260. [Google Scholar] [CrossRef]
- Liu, H.; Baliga, R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Megyesi, J.K.; Kaneto, H.; Price, P.M.; Safirstein, R.L. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. American journal of physiology. Ren. Physiol. 2004, 287, F543–F549. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, H.J.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, J.M. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J. Appl. Toxicol. 2005, 25, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Baba, A.; Matsuo, M.; Itoh, Y.; Oishi, R. Protective effect of cyclic AMP against cisplatin-induced nephrotoxicity. Free Radic. Biol. Med. 2006, 40, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. American journal of physiology. Ren. Physiol. 2005, 289, F166–F174. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int. 2004, 65, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Brian Reeves, W. Cisplatin increases TNF-alpha mRNA stability in kidney proximal tubule cells. Ren. Fail. 2006, 28, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, T.L.; Reed, E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol. Oncol. 1993, 50, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lehane, D.; Winston, A.; Gray, R.; Daskal, Y. The effect of diuretic pre-treatment on clinical, morphological and ultrastructural cis-platinum induced nephrotoxicity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1393–1399. [Google Scholar] [CrossRef]
- Launay-Vacher, V.; Rey, J.B.; Isnard-Bagnis, C.; Deray, G.; Daouphars, M. Prevention of cisplatin nephrotoxicity: State of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother. Pharmacol. 2008, 61, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, J.; Plante, M.; Vergote, I.; du Bois, A.; Hirte, H.; Lacave, A.J.; Wagner, U.; Stahle, A.; Stuart, G.; Kimmig, R. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: An intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [Google Scholar] [CrossRef]
- Lokich, J.; Anderson, N. Carboplatin versus cisplatin in solid tumors: An analysis of the literature. Ann. Oncol. 1998, 9, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Jia, Z.; Sun, Y.; Zhang, A.; Yang, T. A H 2 S Donor GYY4137 Exacerbates Cisplatin-Induced Nephrotoxicity in Mice. Mediat. Inflamm. 2016, 2016, 8145785. [Google Scholar] [CrossRef]
- Cao, X.; Xiong, S.; Zhou, Y.; Wu, Z.; Ding, L.; Zhu, Y.; Wood, M.E.; Whiteman, M.; Moore, P.K.; Bian, J.S. Renal Protective Effect of Hydrogen Sulfide in Cisplatin-Induced Nephrotoxicity. Antioxid. Redox Signal. 2018, 29, 455–470. [Google Scholar] [CrossRef]
- Cao, X.; Bian, J.-S. The Signaling Interaction Systems of in NO Biology and H2S and Medicine. Gasotransmitters 2018, 12, 145. [Google Scholar]
- Cao, X.; Cao, L.; Ding, L.; Bian, J.S. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, Z.; Xiong, S.; Cao, L.; Sethi, G.; Bian, J.S. The role of hydrogen sulfide in cyclic nucleotide signaling. Biochem. Pharmacol. 2018, 149, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, Z.; Cao, X.; Ding, L.; Wen, Z.; Bian, J.S. HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-kappaB and p38 MAPK pathways. Pharmacol. Res. 2016, 111, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Perry, A.; Zhou, Z.; Bucci, M.; Papapetropoulos, A.; Cirino, G.; Wood, M.E. Phosphinodithioate and phosphoramidodithioate hydrogen sulfide donors. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide; Springer: Berlin, Germany, 2015; pp. 337–363. [Google Scholar]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, J.; Tang, C.S.; Geng, B. Effect of synthesized GYY4137, a slowly releasing hydrogen sulfide donor, on cell viability and distribution of hydrogen sulfide in mice. Health Sci. 2010, 42, 493–497. [Google Scholar]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [Green Version]
- Meng, G.; Zhu, J.; Xiao, Y.; Huang, Z.; Zhang, Y.; Tang, X.; Xie, L.; Chen, Y.; Shao, Y.; Ferro, A.; et al. Hydrogen Sulfide Donor GYY4137 Protects against Myocardial Fibrosis. Oxid. Med. Cell. Longev. 2015, 2015, 691070. [Google Scholar] [CrossRef]
- Lin, S.; Visram, F.; Liu, W.; Haig, A.; Jiang, J.; Mok, A.; Lian, D.; Wood, M.E.; Torregrossa, R.; Whiteman, M.; et al. GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J. Urol. 2016, 196, 1778–1787. [Google Scholar] [CrossRef]
- Ikeda, K.; Marutani, E.; Hirai, S.; Wood, M.E.; Whiteman, M.; Ichinose, F. Mitochondria-targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide Biol. Chem. 2015, 49, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H2 S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Maxwell, K.F.; Chrissobolis, S.; Bullen, M.L.; Ku, J.M.; Michael De Silva, T.; Selemidis, S.; Hooker, E.U.; Drummond, G.R.; Sobey, C.G.; et al. Nitroxyl (HNO) suppresses vascular Nox2 oxidase activity. Free Radic. Biol. Med. 2013, 60, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Dusting, G.J.; Peshavariya, H.; Kemp-Harper, B.K.; Drummond, G.R. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 2007, 75, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Ding, L.; Wu, Z.; Cao, X.; Zhang, Q.; Lin, L.; Bian, J.S. Hydrogen sulfide reduces RAGE toxicity through inhibition of its dimer formation. Free Radic. Biol. Med. 2017, 104, 262–271. [Google Scholar] [CrossRef]
- Luan, H.F.; Zhao, Z.B.; Zhao, Q.H.; Zhu, P.; Xiu, M.Y.; Ji, Y. Hydrogen sulfide postconditioning protects isolated rat hearts against ischemia and reperfusion injury mediated by the JAK2/STAT3 survival pathway. Braz. J. Med. Biol. Res. 2012, 45, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Dugbartey, G.J.; Bouma, H.R.; Lobb, I.; Sener, A. Hydrogen sulfide: A novel nephroprotectant against cisplatin-induced renal toxicity. Nitric Oxide 2016, 57, 15–20. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef]
- Kashfi, K. Anti-cancer activity of new designer hydrogen sulfide-donating hybrids. Antioxid. Redox Signal. 2014, 20, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Nie, X.; Xiong, S.; Cao, L.; Wu, Z.; Moore, P.K.; Bian, J.S. Renal protective effect of polysulfide in cisplatin-induced nephrotoxicity. Redox Biol. 2018, 15, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen Sulfide and Polysulfide Signaling. Antioxid. Redox Signal. 2017, 27, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Mukherjea, D.; Jajoo, S.; Kaur, T.; Sheehan, K.E.; Ramkumar, V.; Rybak, L.P. Transtympanic administration of short interfering (si) RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid. Redox Signal. 2010, 13, 589–598. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Kim, S.-J.; Oh, G.S.; Moon, H.-D.; Kwon, K.-B.; Park, C.; Park, B.H.; Lee, H.-K.; Chung, S.-Y. Roles of NADPH oxidases in cisplatin-induced reactive oxygen species generation and ototoxicity. J. Neurosci. 2010, 30, 3933–3946. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Zhang, W.; Moore, P.K.; Bian, J. Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 313. https://doi.org/10.3390/ijms20020313
Cao X, Zhang W, Moore PK, Bian J. Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity. International Journal of Molecular Sciences. 2019; 20(2):313. https://doi.org/10.3390/ijms20020313
Chicago/Turabian StyleCao, Xu, Wencan Zhang, Philip K. Moore, and Jinsong Bian. 2019. "Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity" International Journal of Molecular Sciences 20, no. 2: 313. https://doi.org/10.3390/ijms20020313
APA StyleCao, X., Zhang, W., Moore, P. K., & Bian, J. (2019). Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity. International Journal of Molecular Sciences, 20(2), 313. https://doi.org/10.3390/ijms20020313