Stress Marks on the Genome: Use or Lose?



Abstract
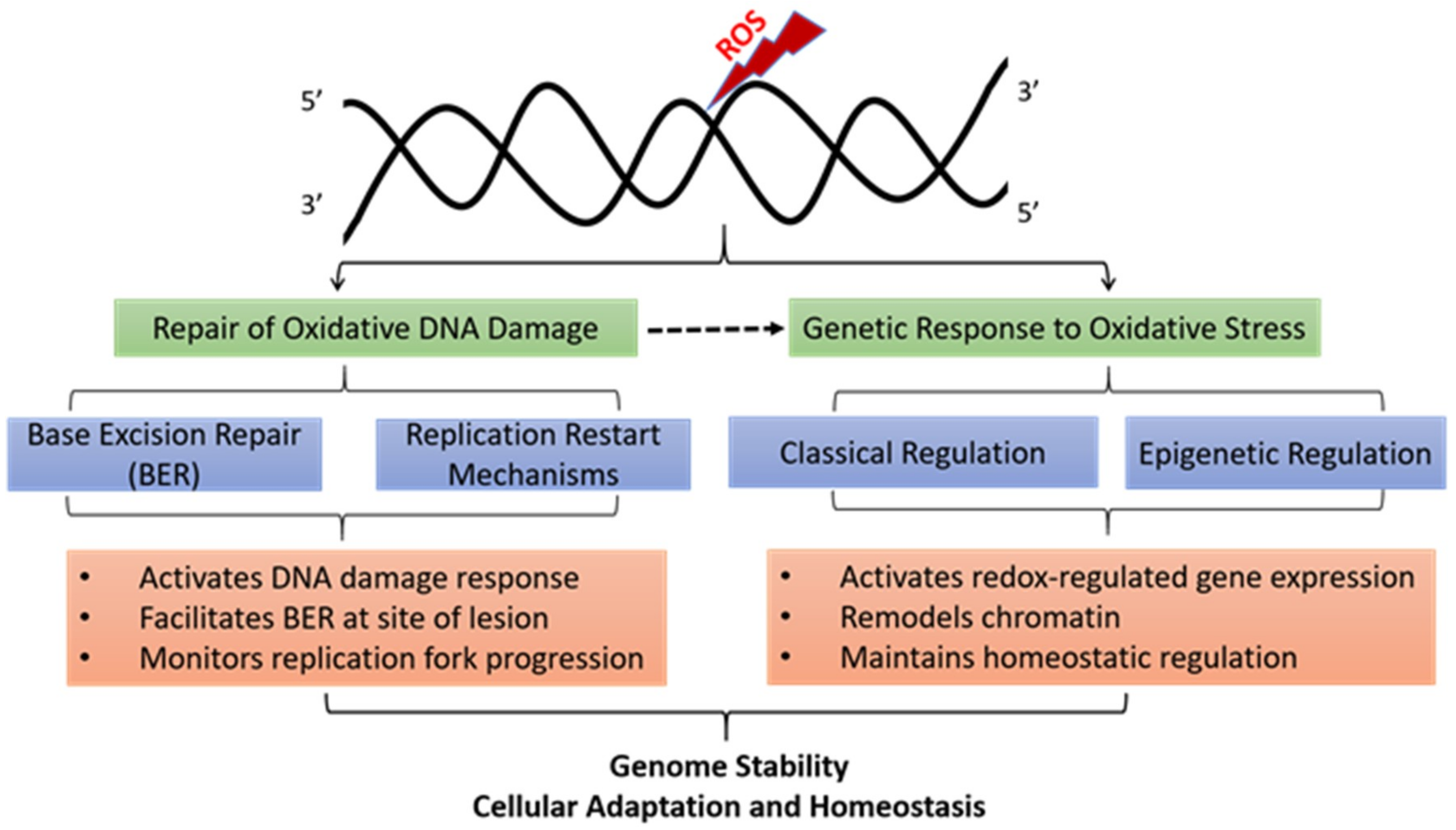
:1. Introduction
2. Specific Oxidative Base Modifications
3. Repair of Oxidative DNA Damage
4. Oxidative DNA Damage and Replication Stress
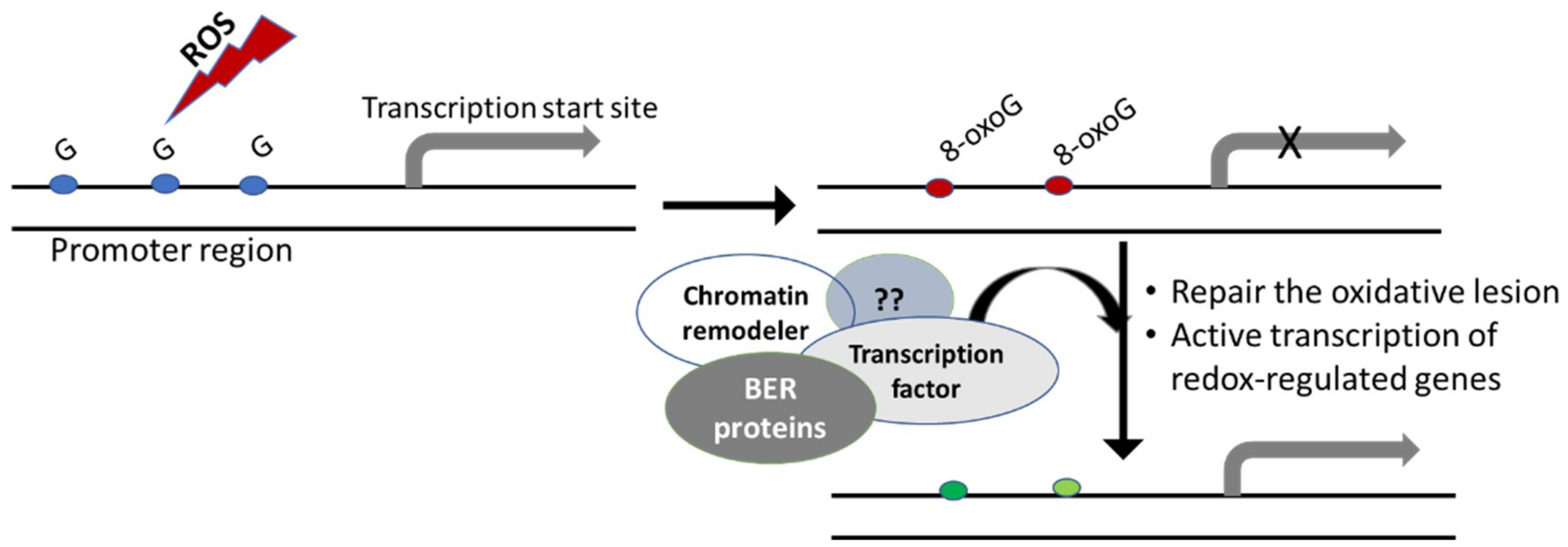
5. Oxidative DNA Damage and Gene Expression Changes
6. Epigenetic Functions of Oxidative DNA Lesions
7. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef]
- Evans, M.D.; Cooke, M.S. Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays 2004, 26, 533–542. [Google Scholar] [CrossRef]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Bohr, V.A.; Stevnsner, T.; de Souza-Pinto, N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002, 286, 127–134. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Batra, V.K.; Wilson, S.H. DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res. 2010, 703, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.J.P.; Hitomi, K.; Tainer, J.A. Flexibility Promotes Fidelity. Structure 2009, 17, 633–634. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Bohr, V.A. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair 2007, 6, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E. Not breathing is not an option: How to deal with oxidative DNA damage. DNA Repair 2017, 59, 82–105. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Odstrcil, E.A.; Tu, B.P.; McKnight, S.L. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science 2007, 316, 1916–1919. [Google Scholar] [CrossRef]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef] [Green Version]
- Klattenhoff, A.W.; Thakur, M.; Chu, C.S.; Ray, D.; Habib, S.L.; Kidane, D. Loss of NEIL3 DNA glycosylase markedly increases replication associated double strand breaks and enhances sensitivity to ATR inhibitor in glioblastoma cells. Oncotarget 2017, 8, 112942–112958. [Google Scholar] [CrossRef]
- Groth, P.; Auslander, S.; Majumder, M.M.; Schultz, N.; Johansson, F.; Petermann, E.; Helleday, T. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol. 2010, 402, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes 2017, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Somyajit, K.; Gupta, R.; Sedlackova, H.; Neelsen, K.J.; Ochs, F.; Rask, M.B.; Choudhary, C.; Lukas, J. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science 2017, 358, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Techer, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [PubMed]
- Sami, F.; Sharma, S. Probing Genome Maintenance Functions of human RECQ. Comput. Struct. Biotechnol. J. 2013, 6, e201303014. [Google Scholar]
- Woodrick, J.; Gupta, S.; Camacho, S.; Parvathaneni, S.; Choudhury, S.; Cheema, A.; Bai, Y.; Khatkar, P.; Erkizan, H.V.; Sami, F.; et al. A new sub-pathway of long-patch base excision repair involving 5′ gap formation. EMBO J. 2017, 36, 1605–1622. [Google Scholar] [CrossRef]
- Sharma, S.; Phatak, P.; Stortchevoi, A.; Jasin, M.; Larocque, J.R. RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair 2012, 11, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [Green Version]
- Parlanti, E.; Locatelli, G.; Maga, G.; Dogliotti, E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007, 35, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Guven, M.; Brem, R.; Macpherson, P.; Peacock, M.; Karran, P. Oxidative Damage to RPA Limits the Nucleotide Excision Repair Capacity of Human Cells. J. Invest. Dermatol. 2015, 135, 2834–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.Y.; Lim, J.W.; Kim, H.; Morio, T.; Kim, K.H. Oxidative Stress Induces Nuclear Loss of DNA Repair Proteins Ku70 and Ku80 and Apoptosis in Pancreatic Acinar AR42J Cells. J. Biol. Chem. 2003, 278, 36676–36687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presence of oxidative stress. Cell Cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Frenk, S.; Houseley, J. Gene expression hallmarks of cellular ageing. Biogerontology 2018, 19, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Costantini, S.; Budillon, A. Oxidative Stress Gene Expression Profile Correlates with Cancer Patient Poor Prognosis: Identification of Crucial Pathways Might Select Novel Therapeutic Approaches. Oxid. Med. Cell. Longev. 2017, 2017, 2597581. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Gibson, G.E. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2007, 21, 276–291. [Google Scholar] [CrossRef]
- Han, E.S.; Muller, F.L.; Perez, V.I.; Qi, W.; Liang, H.; Xi, L.; Fu, C.; Doyle, E.; Hickey, M.; Cornell, J.; et al. The in vivo gene expression signature of oxidative stress. Physiol. Genom. 2008, 34, 112–126. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef] [PubMed]
- Kolbanovskiy, M.; Chowdhury, M.A.; Nadkarni, A.; Broyde, S.; Geacintov, N.E.; Scicchitano, D.A.; Shafirovich, V. The Nonbulky DNA Lesions Spiroiminodihydantoin and 5-Guanidinohydantoin Significantly Block Human RNA Polymerase II Elongation in Vitro. Biochemistry 2017, 56, 3008–3018. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.; Filomeni, G.; Brune, B.; Rotilio, G.; Ciriolo, M.R. Redox mechanisms involved in the selective activation of Nrf2-mediated resistance versus p53-dependent apoptosis in adenocarcinoma cells. J. Biol. Chem. 2009, 284, 27721–27733. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Hajas, G.; Aguilera-Aguirre, L.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Sur, S.; Hazra, T.K.; Mitra, S. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 2012, 287, 20769–20773. [Google Scholar] [CrossRef] [PubMed]
- Seifermann, M.; Epe, B. Oxidatively generated base modifications in DNA: Not only carcinogenic risk factor but also regulatory mark? Free Radic. Biol. Med. 2017, 107, 258–265. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Hosoki, K.; Bacsi, A.; Radak, Z.; Sur, S.; Hegde, M.L.; Tian, B.; Saavedra-Molina, A.; Brasier, A.R.; Ba, X.; et al. Whole transcriptome analysis reveals a role for OGG1-initiated DNA repair signaling in airway remodeling. Free Radic. Biol. Med. 2015, 89, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Qi, T.; Pan, L.; Wang, R.; Zhu, B.; Aguilera-Aguirre, L.; Radak, Z.; Hazra, T.K.; Vlahopoulos, S.A.; Bacsi, A.; et al. Effects of the stimuli-dependent enrichment of 8-oxoguanine DNA glycosylase1 on chromatinized DNA. Redox Biol. 2018, 18, 43–53. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, C.; Qiao, P.; Xue, Y.; Zheng, X.; Chen, H.; Zeng, X.; Liu, W.; Boldogh, I.; Ba, X. OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos. Cell. Death Dis. 2018, 9, 628. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009, 11, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Lirussi, L.; D’Ambrosio, C.; Dal Piaz, F.; Vascotto, C.; Casarano, E.; Marasco, D.; Scaloni, A.; Fogolari, F.; Tell, G. SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol. Biol. Cell 2014, 25, 532–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastukh, V.; Roberts, J.T.; Clark, D.W.; Bardwell, G.C.; Patel, M.; Al-Mehdi, A.B.; Borchert, G.M.; Gillespie, M.N. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1367–L1375. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef]
- Moore, S.P.; Toomire, K.J.; Strauss, P.R. DNA modifications repaired by base excision repair are epigenetic. DNA Repair 2013, 12, 1152–1158. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [Green Version]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef]
- Sun, D.; Liu, W.J.; Guo, K.; Rusche, J.J.; Ebbinghaus, S.; Gokhale, V.; Hurley, L.H. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol. Cancer Ther. 2008, 7, 880–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Menezo, Y.J.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ji, D.; Lin, K.; Song, J.; Wang, Y. Effects of Tet-induced oxidation products of 5-methylcytosine on Dnmt1- and DNMT3a-mediated cytosine methylation. Mol. Biosyst. 2014, 10, 1749–1752. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef]
- Chen, G.; Chen, J.; Yan, Z.; Li, Z.; Yu, M.; Guo, W.; Tian, W. Maternal diabetes modulates dental epithelial stem cells proliferation and self-renewal in offspring through apurinic/apyrimidinicendonuclease 1-mediated DNA methylation. Sci. Rep. 2017, 7, 40762. [Google Scholar] [CrossRef] [Green Version]
- Bravard, A.; Vacher, M.; Gouget, B.; Coutant, A.; de Boisferon, F.H.; Marsin, S.; Chevillard, S.; Radicella, J.P. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol. Cell. Biol. 2006, 26, 7430–7436. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Campalans, A.; Vacher, M.; Gouget, B.; Levalois, C.; Chevillard, S.; Radicella, J.P. Inactivation by oxidation and recruitment into stress granules of hOGG1 but not APE1 in human cells exposed to sub-lethal concentrations of cadmium. Mutat. Res. 2010, 685, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006, 26, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Hinz, J.M.; Czaja, W. Facilitation of base excision repair by chromatin remodeling. DNA Repair 2015, 36, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Balliano, A.J.; Hayes, J.J. Base excision repair in chromatin: Insights from reconstituted systems. DNA Repair 2015, 36, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Fedeles, B.I. G-quadruplex-forming promoter sequences enable transcriptional activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, 2788–2790. [Google Scholar] [CrossRef]
- Li, X.L.; Lu, X.; Parvathaneni, S.; Bilke, S.; Zhang, H.; Thangavel, S.; Vindigni, A.; Hara, T.; Zhu, Y.; Meltzer, P.S.; et al. Identification of RECQ1-regulated transcriptome uncovers a role of RECQ1 in regulation of cancer cell migration and invasion. Cell Cycle 2014, 13, 2431–2445. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Parvathaneni, S.; Li, X.L.; Lal, A.; Sharma, S. Transcriptome guided identification of novel functions of RECQ1 helicase. Methods 2016, 108, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.H.; Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Welsh, J.A.; Schetter, A.J.; Kumamoto, K.; Wang, X.W.; Hickson, I.D.; et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc. Natl. Acad. Sci. USA 2014, 111, 9905–9910. [Google Scholar] [CrossRef]
- Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Nguyen, G.H.; Oshima, J.; Maizels, N.; Harris, C.C.; Monnat, R.J. Werner syndrome helicase modulates G4 DNA-dependent transcription and opposes mechanistically distinct senescence-associated gene expression programs. bioRxiv 2015. [Google Scholar] [CrossRef]
- Popuri, V.; Bachrati, C.Z.; Muzzolini, L.; Mosedale, G.; Costantini, S.; Giacomini, E.; Hickson, I.D.; Vindigni, A. The Human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 2008, 283, 17766–17776. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.W.; Cattoglio, C.; Tjian, R. The Intertwined Roles of Transcription and Repair Proteins. Mol. Cell 2013, 52, 291–302. [Google Scholar] [CrossRef]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| DNA Base | Oxidized Base Modification |
|---|---|
| Guanine (G) | 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxoG) 8-oxoG is further oxidized to: Spiroiminodihydantoin Guanidinohydantoin 2,6-diamino-4-hydroxy-5 formamidopyrimidine (FapyG) |
| Cytosine (C) | 5-hydroxy-2′-deoxycytidine (OH5C) |
| Adenine (A) | 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxoA) 4,6-diamino-5-formamidopyrimidine (FapyA) 2-hydroxydeoxyadenosine-5′-triphosphate (2OHA) |
| Thymine (T) | Thymine glycol (Tg) 5,6-dihydrothymine (DHT) 5-hydroxymethyluracil (5hmU) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bokhari, B.; Sharma, S. Stress Marks on the Genome: Use or Lose? Int. J. Mol. Sci. 2019, 20, 364. https://doi.org/10.3390/ijms20020364
Bokhari B, Sharma S. Stress Marks on the Genome: Use or Lose? International Journal of Molecular Sciences. 2019; 20(2):364. https://doi.org/10.3390/ijms20020364
Chicago/Turabian StyleBokhari, Bayan, and Sudha Sharma. 2019. "Stress Marks on the Genome: Use or Lose?" International Journal of Molecular Sciences 20, no. 2: 364. https://doi.org/10.3390/ijms20020364
APA StyleBokhari, B., & Sharma, S. (2019). Stress Marks on the Genome: Use or Lose? International Journal of Molecular Sciences, 20(2), 364. https://doi.org/10.3390/ijms20020364