Comprehensive Analysis of Differentially Expressed Unigenes under NaCl Stress in Flax (Linum usitatissimum L.) Using RNA-Seq

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Response of Flax to NaCl Stress
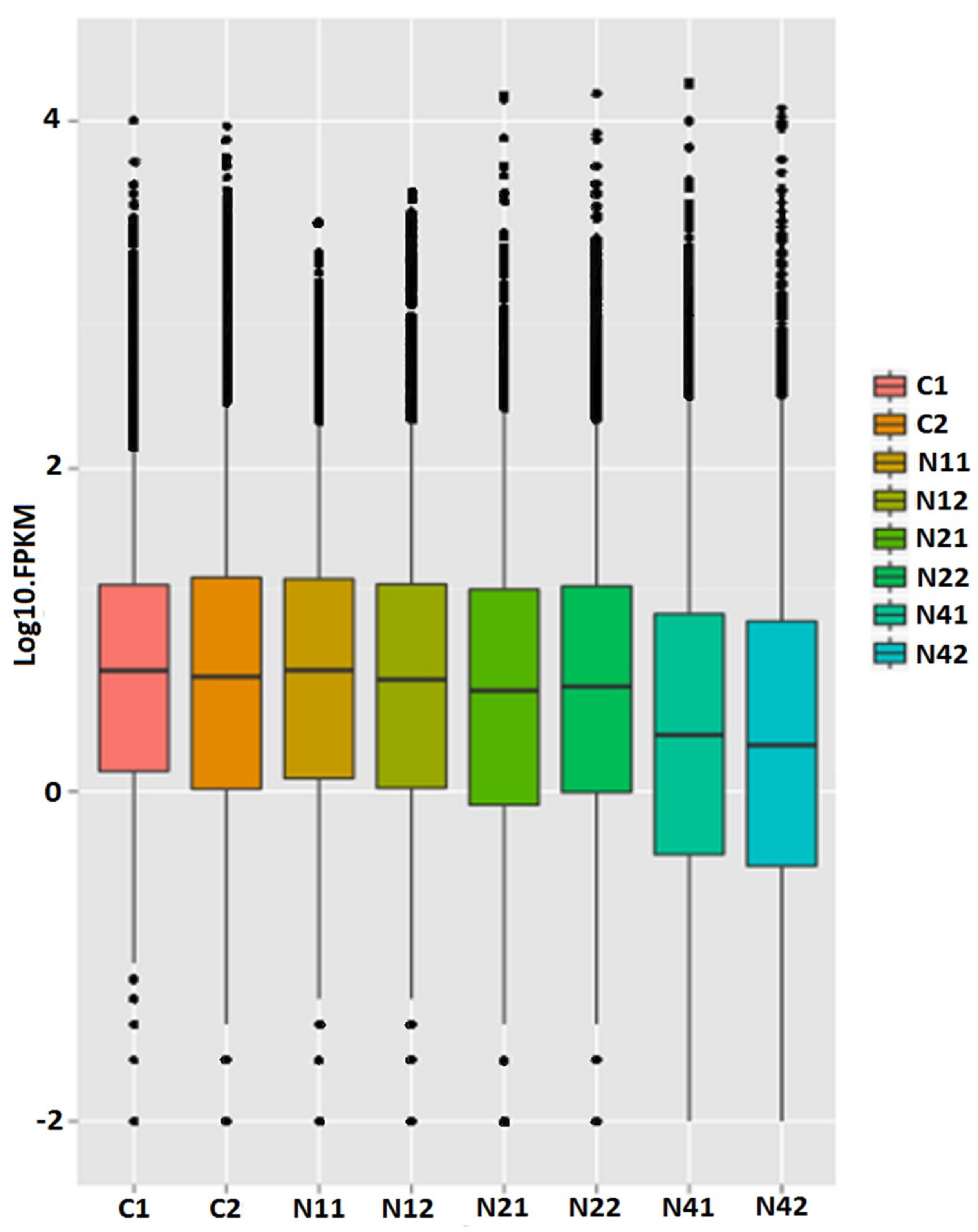
2.2. Transcriptome Analysis
2.3. Identification of Differentially Expressed Unigenes (DEUs)
2.4. Cluster Analysis of DEUs
2.5. Functional Annotation
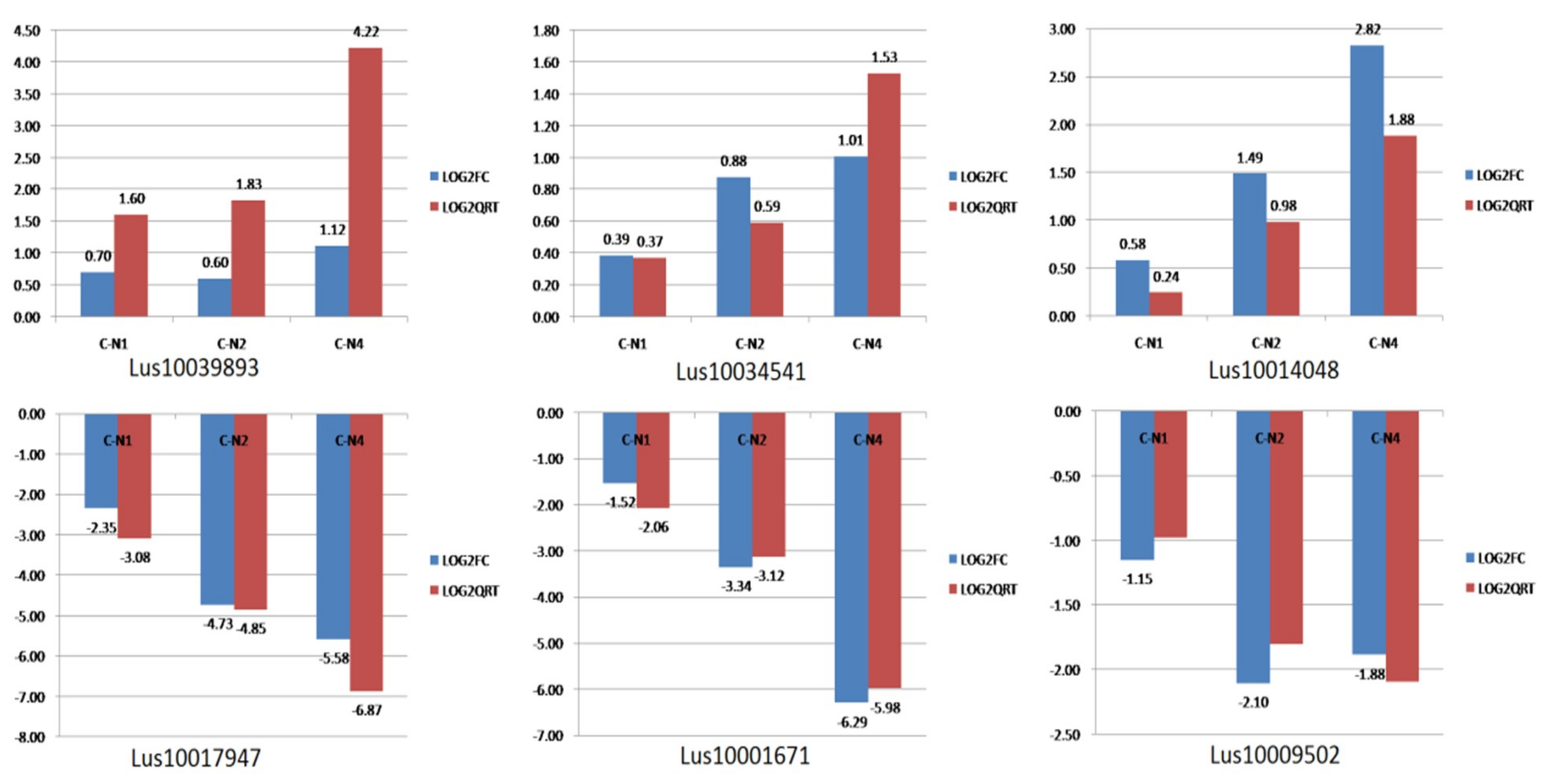
2.6. RNA-Seq Expression Validation
2.7. Distribution Characteristics of EST-SSRs
3. Materials and Methods
3.1. Material Planting and Processing
3.2. Stress Treatments and Sample Preparation
3.3. RNA Isolation and cDNA Library Construction
3.4. Illumina Sequencing, Assembly and Annotation
3.5. Identification of DEUs and Cluster Analysis
3.6. Real-Time qRT-PCR Validation
3.7. Development of EST-SSR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, Y.; Huang, W.G.; Chen, H.Y.; Wu, G.W.; Yuan, H.M.; Song, X.X.; Kang, Q.H.; Zhao, D.S.; Jiang, W.D.; Liu, Y.; et al. Identification of differentially expressed genes in flax (Linum usitatissimum L.) under saline-alkaline stress by digital gene expression. Gene 2014, 549, 113–122. [Google Scholar] [CrossRef] [PubMed]
- El-Hariri, D.M.; Al-Kordy, M.A.; Hassanein, M.S.; Ahmed, M.A. Partition of photosynthates and energy production in different flax cultivars. J. Nat. Fibers 2005. [Google Scholar] [CrossRef]
- Pecenka, R.; Fürll, C.; Ola, D.C.; Budde, J.; Gusovius, H.J. Efficient use of agricultural land in production of energy: Natural insulation versus bio-energy. In Proceedings of the International Conference of Agricultural Engineering CIGR-AgEng, Agriculture and Engineering for a Healthier Life, Valencia, Spain, 8–12 July 2012. [Google Scholar]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G. Flax: Breeding and utilisation. Plant Growth Regul. 1991, 10, 171–172. [Google Scholar] [CrossRef]
- Abdel, W.W.; Ahmed, S.A. Response surface methodology for production, characterization and application of solvent, salt and alkali-tolerant alkaline protease from isolated fungal strain, Aspergillus niger, WA 2017. Int. J. Biol. Macromol. 2018, 115, 447–458. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and Drought Stress Signal Transduction in Plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef]
- Zhao, G.; Zhao, Y.Y.; Yu, X.L.; Kiprotich, F.; Han, H.; Guan, R.Z.; Wang, R.; Shen, W.B. Nitric Oxide Is Required for Melatonin-Enhanced Tolerance against Salinity Stress in Rapeseed (Brassica napus L.) Seedlings. Int. J. Mol. Sci. 2018, 19, 1912. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Bulbul, S.; Piao, W.; Choi, G.; Paek, N.C. Arabidopsis EARLY FLOWERING3 increases salt tolerance by suppressing salt stress response pathways. Plant J. 2017, 92, 1106–1120. [Google Scholar] [CrossRef]
- Li, Y. Effect of salt stress on seed germination and seedling growth of three salinity plants. Pak. J. Biol. Sci. 2008, 11, 1268–1272. [Google Scholar] [CrossRef]
- Çavuşoğlu, K.; Kılıç, S.; Kabar, K. Some morphological and anatomical observations during alleviation of salinity (NaCl) stress on seed germination and seedling growth of barley by polyamines. Acta Physiol. Plant. 2007, 29, 551–557. [Google Scholar] [CrossRef]
- Jiang, A.; Gan, L.; Tu, Y.; Ma, H.; Zhang, J.; Song, Z.; He, Y.C.; Cai, D.T.; Xue, X.D. The effect of genome duplication on seed germination and seedling growth of rice under salt stress. Aust. J. Crop Sci. 2013, 7, 1814–1821. [Google Scholar]
- François, T.; Parent, B.; Caldeira, C.F.; Welcker, C. Genetic and physiological controls of growth under water deficit. Plant Physiol. 2014, 164, 1628–1635. [Google Scholar] [CrossRef]
- Álvarez, S.; Rodríguez, P.; Broetto, F.; Sánchez-Blanco, M.J. Long term responses and adaptive strategies of pistacialentiscus under moderate and severe deficit irrigation and salinity: Osmotic and elastic adjustment, growth, ion uptake and photosynthetic activity. Agric. Water Manag. 2018, 202, 253–262. [Google Scholar] [CrossRef]
- Li, H.; Chang, J.; Chen, H.; Wang, Z.; Gu, X.; Wei, C.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X. Exogenous melatonin confers salt stress tolerance to watermelon by improving photosynthesis and redox homeostasis. Front. Plant Sci. 2017, 8, 295. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Yang, L.E.; Fletcher, N.K.; Lee, D.H.; Kocinsky, H.; Bachmann, S.; Delpire, E.; McDonough, A. Effects of K+-deficient diets with and without nacl supplementation on Na+, K+, and H2O transporters’ abundance along the nephron. Am. J. Physiol. Renal Physiol. 2012, 303, F92–F104. [Google Scholar] [CrossRef] [PubMed]
- Rus, A.; Yokoi, S.; Sharkhuu, A.; Reddy, M.; Lee, B.H.; Matsumoto, T.K.; Koiwa, H.; Zhu, J.K.; Bressan, R.A.; Hasegawa, P.M. Athkt1 is a salt tolerance determinant that controls Na+ entry into plant roots. Proc. Natl. Acad. Sci. USA 2001, 98, 14150–14155. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Lilley, C.J.; Urwin, P.E. Identification of genes involved in the response of Arabidopsis to simultaneous biotic and abiotic stresses. Plant Physiol. 2013, 162, 2028–2041. [Google Scholar] [CrossRef]
- Han, N.; Lan, W.J.; He, X.; Shao, Q.; Wang, B.S.; Zhao, X.J. Expression of a Suaeda salsa Vacuolar H+/Ca2+ Transporter Gene in Arabidopsis Contributes to Physiological Changes in Salinity. Plant Mol. Boil. Rep. 2012, 30, 470–477. [Google Scholar] [CrossRef]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Wei, L.; Qiang, Z.; Qi, F.; Yan, Z.; Guo, Y.; Li, W. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome Analysis of Salt Stress Responsiveness in the Seedlings of Dongxiang Wild Rice (Oryza rufipogon Griff.). PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Xiao, Z.; Li, M.W.; Wong, F.L.; Yung, W.S.; Ku, Y.S.; Wang, Q.; Wang, X.; Xie, M.; Yim, A.K. Transcriptomic reprogramming in soybean seedlings under salt stress. Plant Cell Environ. 2018, 12, e0189159. [Google Scholar] [CrossRef]
- Guo, J.; Li, Y.; Han, G.; Song, J.; Wang, B. NaCl markedly improved the reproductive capacity of the euhalophyte Suaeda Salsa. Funct. Plant Boil. 2018, 45, 350. [Google Scholar] [CrossRef]
- Cui, F.; Sui, N.; Duan, G.; Liu, Y.; Han, Y.; Liu, S.; Wan, S.; Li, G. Identification of Metabolites and Transcripts Involved in Salt Stress and Recovery in Peanut. Front. Plant Sci. 2018, 9, 217. [Google Scholar] [CrossRef]
- Yuan, F.; Lyu, M.J.A.; Leng, B.Y.; Zhu, X.G.; Wang, B.S. The transcriptome of NaCl-treated Limonium bicolor leaves reveals the genes controlling salt secretion of salt gland. Plant Mol. Boil. 2016, 91, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Qian, Z.; Luo, M.; Fan, S.; Zhang, X.; Zhang, L. Identification of salt stress responding genes using transcriptome analysis in green alga chlamydomonas reinhardtii. Int. J. Mol. Sci. 2018, 19, 3359. [Google Scholar] [CrossRef]
- Li, H.; Lin, J.; Yang, Q.S.; Li, X.G.; Chang, Y.H. Comprehensive analysis of differentially expressed genes under salt stress in pear (Pyrus betulaefolia) using RNA-seq. Plant Growth Regul. 2017, 82, 409–420. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Li, X.G.; Lin, J.; Wang, Z.H.; Yang, Q.S.; Chang, Y.H. Transcriptome sequencing and analysis of major genes involved in calcium signaling pathways in pear plants (Pyrus calleryana Decne.). BMC Genom. 2015, 16, 738. [Google Scholar] [CrossRef]
- Hamed, K.B.; Ellouzi, H.; Talbi, O.Z.; Hessini, K.; Slama, I.; Ghnaya, T.; Bosch, S.M.; Savour, A.; Abdelly, C. Physiological response of halophytes to multiple stresses. Funct. Plant Biol. 2013, 40, 883–896. [Google Scholar] [CrossRef]
- Dash, P.K.; Cao, Y.; Jailani, A.K.; Gupta, P.; Venglat, P.; Xiang, D.; Rai, R.; Sharma, R.; Thirunavukkarasu, N.; Abdin, M.Z.; et al. Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). Gm Crop. Food 2014, 5, 106–119. [Google Scholar] [CrossRef]
- Dewey, C.N.; Bo, L. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Antónia, G.; David, R.; Matczuk, K.; Nikoleta, D.; David, C.; Twell, D.; Honys, D. AtbZIP34 is required for Arabidopsis pollen wall patterning and the control of several metabolic pathways in developing pollen. Plant Mol. Biol. 2009, 70, 581–601. [Google Scholar] [CrossRef]
- Miao, H.X.; Sun, P.G.; Liu, J.H.; Wang, J.Y.; Xu, B.Y.; Jin, Z.Q. Overexpression of a Novel ROP Gene from the Banana (MaROP5g) Confers Increased Salt Stress Tolerance. Int. J. Mol. Sci. 2018, 19, 3108. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, Q.; Sun, D.; Wu, G.; Zhang, L.; Yuan, H.; Yu, Y.; Zhang, S.; Yang, X.; Li, Z.; et al. Transcriptome analysis of flax (Linum usitatissimum L.) undergoing osmotic stress. Ind. Crop. Prod. 2018, 116, 215–223. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2014, 32, D258–D261. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * | Clean Reads | Total Mapped | Unique Mapped | Multi Mapped | Bases (Gb) | Q20 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| C1 | 60,125,334 | 25,807,774 | 9,037,386 | 16,770,388 | 7.48 | 99.36 | 48.35 |
| C2 | 54,128,778 | 23,792,226 | 9,600,424 | 14,191,802 | 6.67 | 99.23 | 47.68 |
| N11 | 62,422,020 | 26,632,880 | 9,637,906 | 16,994,974 | 7.77 | 99.33 | 47.67 |
| N12 | 57,381,976 | 22,307,914 | 8,055,084 | 14,252,830 | 7.13 | 99.3 | 47.84 |
| N21 | 60,066,230 | 25,957,230 | 9,698,022 | 16,259,208 | 7.48 | 99.37 | 47.87 |
| N22 | 21,827,146 | 9,277,362 | 3,136,434 | 6,140,928 | 2.58 | 97.64 | 47.81 |
| N41 | 56,385,726 | 25,227,542 | 9,202,726 | 16,024,816 | 7.01 | 99.35 | 47.96 |
| N42 | 59,471,292 | 26,454,904 | 9,667,984 | 16,786,920 | 7.39 | 99.31 | 48.03 |
| FPKM Interval | 0–0.1 | 0.1–1 | 1–3 | 3–15 | 15–60 | >60 |
|---|---|---|---|---|---|---|
| C1 | 1635 (3.03%) | 9893 (18.31%) | 11,852 (21.93%) | 21,176 (39.19%) | 7660 (14.17%) | 1823 (3.37%) |
| C2 | 1805 (3.37%) | 11,354 (21.21%) | 11,228 (20.97%) | 18,620 (34.78%) | 8258 (15.42%) | 2278 (4.25%) |
| N11 | 1805 (3.31%) | 10,645 (19.52%) | 11,210 (20.56%) | 20,619 (37.82%) | 8343 (15.30%) | 1901 (3.49%) |
| N12 | 1784 (3.28%) | 11,534 (21.18%) | 11,789 (21.65%) | 19,438 (35.70%) | 7855 (14.43%) | 2047 (3.76%) |
| N21 | 2226 (4.15%) | 12,475 (23.27%) | 11,564 (21.57%) | 17,954 (33.49%) | 7324 (13.66%) | 2061 (3.84%) |
| N22 | 1015 (2.03%) | 11,621 (23.24%) | 11,373 (22.75%) | 16,902 (33.80%) | 7049 (14.10%) | 2039 (4.08%) |
| N41 | 3221 (6.32%) | 15,996 (31.38%) | 10,872 (21.33%) | 13,032 (25.57%) | 5696 (11.17%) | 2154 (4.23%) |
| N42 | 3804 (7.43%) | 16,694 (32.61%) | 10,687 (20.88%) | 12,463 (24.35%) | 5431 (10.61%) | 2111 (4.12%) |
| Type | Repeat Number | Percentage (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | >10 | Total | ||
| Dinucleotide | 238 | 148 | 82 | 57 | 38 | 60 | 623 | 35.06 | ||
| Trinucleotide | 558 | 250 | 107 | 49 | 36 | 2 | 1002 | 56.39 | ||
| Quadnucleotide | 53 | 12 | 3 | 68 | 3.83 | |||||
| Pentanucleotide | 49 | 7 | 2 | 58 | 3.26 | |||||
| Hexanucleotide | 23 | 3 | 26 | 1.46 | ||||||
| Total | 72 | 621 | 502 | 258 | 131 | 93 | 40 | 60 | 1777 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Zhao, Q.; Wu, G.; Yuan, H.; Ma, Y.; Lin, H.; Pan, L.; Li, S.; Sun, D. Comprehensive Analysis of Differentially Expressed Unigenes under NaCl Stress in Flax (Linum usitatissimum L.) Using RNA-Seq. Int. J. Mol. Sci. 2019, 20, 369. https://doi.org/10.3390/ijms20020369
Wu J, Zhao Q, Wu G, Yuan H, Ma Y, Lin H, Pan L, Li S, Sun D. Comprehensive Analysis of Differentially Expressed Unigenes under NaCl Stress in Flax (Linum usitatissimum L.) Using RNA-Seq. International Journal of Molecular Sciences. 2019; 20(2):369. https://doi.org/10.3390/ijms20020369
Chicago/Turabian StyleWu, Jianzhong, Qian Zhao, Guangwen Wu, Hongmei Yuan, Yanhua Ma, Hong Lin, Liyan Pan, Suiyan Li, and Dequan Sun. 2019. "Comprehensive Analysis of Differentially Expressed Unigenes under NaCl Stress in Flax (Linum usitatissimum L.) Using RNA-Seq" International Journal of Molecular Sciences 20, no. 2: 369. https://doi.org/10.3390/ijms20020369
APA StyleWu, J., Zhao, Q., Wu, G., Yuan, H., Ma, Y., Lin, H., Pan, L., Li, S., & Sun, D. (2019). Comprehensive Analysis of Differentially Expressed Unigenes under NaCl Stress in Flax (Linum usitatissimum L.) Using RNA-Seq. International Journal of Molecular Sciences, 20(2), 369. https://doi.org/10.3390/ijms20020369