Analytical Methods for Detection of Plant Metabolomes Changes in Response to Biotic and Abiotic Stresses



Abstract
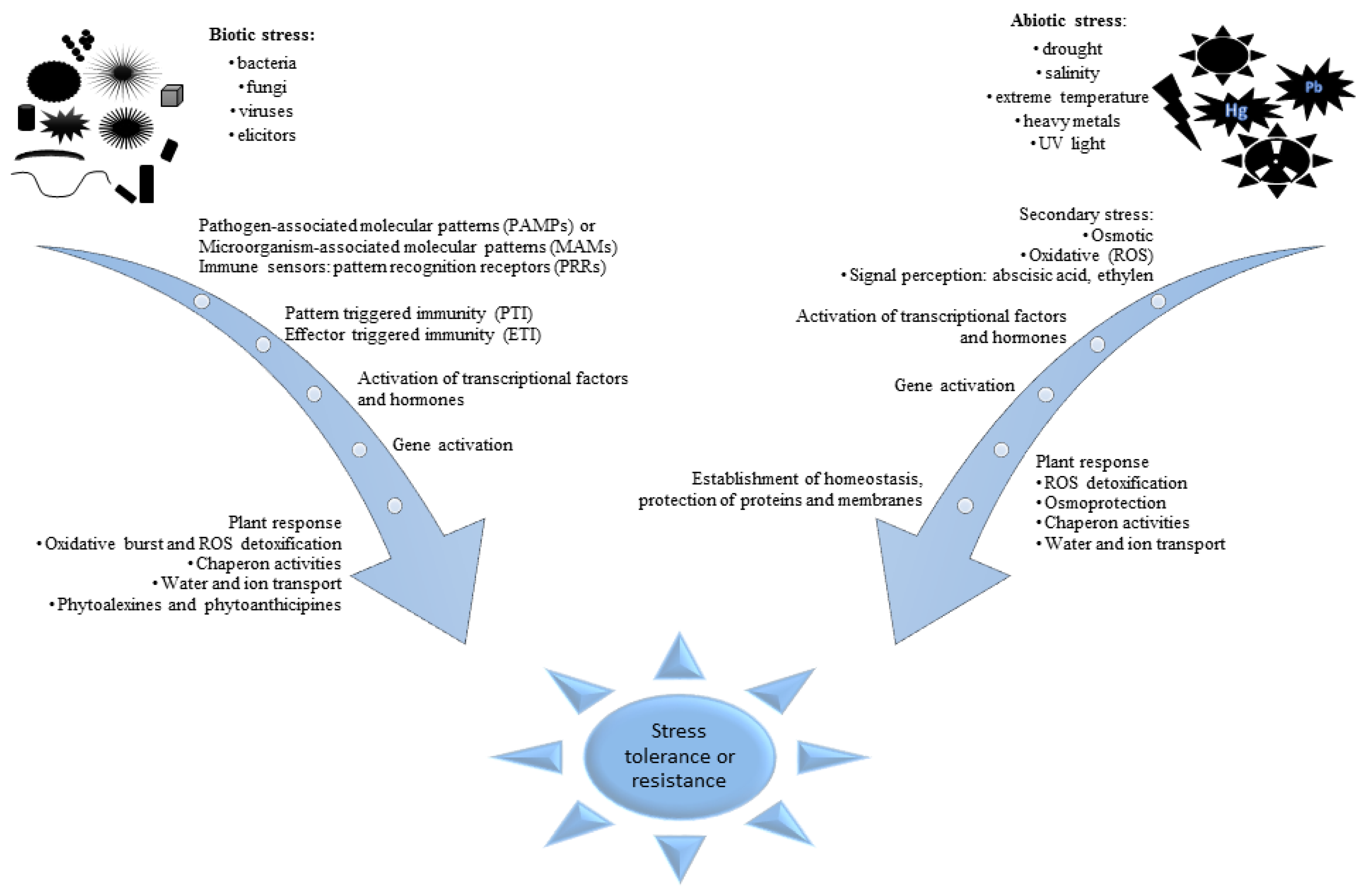
:1. Introduction
2. Plants and Biotic and/or Abiotic Stress
3. Analytical Methods Applied for Metabolome Composition Analysis and Description
4. Bioinformatics and Statistical Analysis in Metabolomics
4.1. Tools and Software Dedicated to Metabolomics
4.2. Peak Picking
4.3. Data Reduction
4.4. Data Set Alignment
4.5. Post-Processing and Statistic of Data Table
4.6. Visualization of Statistical Results
4.7. Metabolomics Enhancement
Funding
Conflicts of Interest
References
- Lobell, D.B.; Field, C.B. Global scale climate–crop yield relationships and the impacts of recent warming. Environ. Res. Lett. 2007, 2, 014002. [Google Scholar] [CrossRef] [Green Version]
- Lobell, D.B.; Gourdji, S.M. The Influence of Climate Change on Global Crop Productivity. Plant Physiol. 2012, 160, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, A.C.; Castroverde, C.D.M.; He, S.Y. Plant–Pathogen Warfare under Changing Climate Conditions. Curr. Biol. 2018, 28, R619–R634. [Google Scholar] [CrossRef]
- Dawid, C.; Hille, K. Functional Metabolomics—A Useful Tool to Characterize Stress-Induced Metabolome Alterations Opening New Avenues towards Tailoring Food Crop Quality. Agronomy 2018, 8, 138. [Google Scholar] [CrossRef]
- Christ, B.; Pluskal, T.; Aubry, S.; Weng, J.K. Contribution of Untargeted Metabolomics for Future Assessment of Biotech Crops. Trends Plant Sci. 2018, 23, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Saito, K. Phytochemical genomics—A new trend. Curr. Opin. Plant Biol. 2013, 16, 373–380. [Google Scholar] [CrossRef]
- Fuhrer, T.; Zamboni, N. High-throughput discovery metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Quanbeck, S.M.M.; Brachova, L.; Campbell, A.A.; Guan, X.; Perera, A.; He, K.; Rhee, S.Y.; Bais, P.; Dickerson, J.A.; Wohlgemuth, G.; et al. Metabolomics as a hypothesis-generating functional genomics tool for the annotation of Arabidopsis thaliana genes of “unknown function”. Front. Plant Sci. 2012, 3, 15. [Google Scholar] [CrossRef]
- Hartmann, T. From waste products to ecochemicals: Fifty years research of plant secondary metabolism. Phytochemistry 2007, 68, 2831–2846. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Hankemeier, T.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viant, M.R.; Kurland, J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Rollinger, J.M.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cai, X. Climate change impacts on global agricultural land availability. Environ. Res. Lett. 2011, 6, 014014. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarek, P. Chemical warfare or modulators of defence responses—The function of secondary metabolites in plant immunity. Curr. Opin. Chem. Biol. 2012, 15, 407–414. [Google Scholar] [CrossRef]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling Mechanisms in Pattern-Triggered Immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Misra, B.B.; de Armas, E.; Chen, S. Differential metabolomic responses of PAMP-triggered immunity and effector-triggered immunity in Arabidopsis suspension cells. Metabolomics 2016, 12, 61. [Google Scholar] [CrossRef]
- Boutrot, F.; Zipfel, C. Function, Discovery, and Exploitation of Plant Pattern Recognition Receptors for Broad-Spectrum Disease Resistance. Ann. Rev. Phytopathol. 2017, 55, 257–286. [Google Scholar] [CrossRef]
- Ranf, S. Pattern Recognition Receptors—Versatile Genetic Tools for Engineering Broad-Spectrum Disease Resistance in Crops. Agronomy 2018, 8, 134. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Saito, K. Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Chem. Biol. 2015, 24, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, M.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- Lawlor, D.W. Genetic engineering to improve plant performance under drought: Physiological evaluation of achievements, limitations and possibilities. J. Exp. Bot. 2013, 64, 83–108. [Google Scholar] [CrossRef]
- Tester, M.; Bacic, A. Abiotic Stress Tolerance in Grasses. From Model Plants to Crop Plants. Plant Physiol. 2005, 137, 791–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noctor, G.; Mhamdi, A.; Foyer, C.H. The Roles of Reactive Oxygen Metabolism in Drought: Not So Cut and Dried. Plant Physiol. 2014, 164, 1636–1648. [Google Scholar] [CrossRef] [Green Version]
- Fahad, S.; Bajwa, A.A.; Nazir, U.; Anjum, S.A.; Farooq, A.; Zohaib, A.; Sadia, S.; Nasim, W.; Adkins, A.; Saud, S.; et al. Crop Production under Drought and Heat Stress: Plant Responses and Management Options. Front. Plant Sci. 2017, 8, 1147. [Google Scholar] [CrossRef]
- Bechtold, U. Plant Life in Extreme Environments: How Do You Improve Drought Tolerance? Front. Plant Sci. 2018, 9, 543. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3544. [Google Scholar] [CrossRef]
- Nam, M.H.; Bang, E.; Kwon, T.Y.; Kim, Y.; Kim, E.H.; Cho, K.; Park, W.J.; Kim, B.-G.; Yoon, I.S. Metabolite Profiling of Diverse Rice Germplasm and Identification of Conserved Metabolic Markers of Rice Roots in Response to Long-Term Mild Salinity Stress. Int. J. Mol. Sci. 2015, 16, 21959–21974. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, K.G.; Kushalappa, A.C.; Choo, T.M.; Dion, Y.; Rioux, S. Mass spectrometry based metabolomics to identify potential biomarkers for resistance in barley against fusarium head blight (Fusarium graminearum). J. Chem. Ecol. 2011, 37, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Bollina, V.; Kushalappa, A.C.; Choo, T.M.; Dion, Y.; Rioux, S. Identification of metabolites related to mechanisms of resistance in barley against Fusarium graminearum, based on mass spectrometry. Plant Mol. Biol. 2011, 77, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Sawikowska, A.; Kuczyńska, A.; Ogrodowicz, P.; Mikołajczak, K.; Krystkowiak, K.; Gudyś, K.; Guzy-Wróbelska, J.; Krajewski, P.; Kachlicki, P. Drought related secondary metabolites of barley (Hordeum vulgare L.) leaves and their mQTLs. Plant J. 2017, 89, 898–913. [Google Scholar] [CrossRef] [PubMed]
- Kleinwächter, M.; Selmar, D. New insights explain that drought stress enhances the quality of spice and medicinal plants: Potential applications. Agron. Sustain. Dev. 2015, 35, 121–131. [Google Scholar] [CrossRef]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Shukla, P.S.; Gupta, K.; Jha, B. Bioengineering for Salinity Tolerance in Plants: State of the Art. Mol. Biotechnol. 2013, 54, 102–123. [Google Scholar] [CrossRef]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for Plant Improvement: Status and Prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef]
- Tang, X.; Mu, X.; Shao, H.; Wang, H.; Brestic, M. Global plant-responding mechanisms to salt stress: Physiological and molecular levels and implications in biotechnology. Crit. Rev. Biotechnol. 2015, 35, 425–437. [Google Scholar] [CrossRef]
- Kusano, M.; Saito, K. Role of Metabolomics in Crop Improvement. J. Plant Biochem. Biotechnol. 2012, 21, S24–S31. [Google Scholar] [CrossRef]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2008, 25, 39–48. [Google Scholar] [CrossRef]
- Saxena, A.; Cramer, C.S. Metabolomics: A Potential Tool for Breeding Nutraceutical Vegetables. Adv. Crop Sci. Technol. 2013, 1, 106. [Google Scholar] [CrossRef]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant Metabolomics: An Indispensable System Biology Tool for Plant Science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef]
- Farrant, J.M.; Cooper, K.; Hilgart, A.; Abdalla, K.O.; Bentley, J.; Thomson, J.A.; Dace, H.J.W.; Peton, N.; Mundree, S.G.; Rafudeen, S.M. A molecular physiological review of vegetative desiccation tolerance in the resurrection plant Xerophyta viscosa (Baker). Planta 2015, 242, 407–426. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, R.; Saito, K. Metabolomics for unknown plant metabolites. Anal. Bioanal. Chem. 2013, 405, 5005–5011. [Google Scholar] [CrossRef]
- Bayer, E.; Albert, K.; Nieder, M.; Grome, E.; Wolff, G.; Rindlisbacher, M. Online coupling of liquid chromatography and high field nuclear magnetic resonance spectrometry. Anal. Chem. 1982, 54, 1747–1750. [Google Scholar] [CrossRef]
- Jaroszewski, J.W. Hyphenated NMR methods in natural products research, part 1: Direct hyphenation. Planta Medica 2005, 71, 691–700. [Google Scholar] [CrossRef]
- Jaroszewski, J.W. Hyphenated NMR methods in natural products research, Part 2: HPLC-SPE-NMR and other new trends in NMR hyphenation. Planta Medica 2005, 71, 795–802. [Google Scholar] [CrossRef]
- Prinsloo, G.; Vervoort, J. Identifying anti-HSV compounds from unrelated plants using NMR and LC–MS metabolomic analysis. Metabolomics 2018, 14, 134. [Google Scholar] [CrossRef]
- Hou, S.; Zhu, J.; Ding, M.; Lv, G. Simultaneous determination of gibberellic acid, indole-3-acetic acid and abscisic acid in wheat extracts by solid-phase extraction and liquid chromatography–electrospray tandem mass spectrometry. Talanta 2008, 76, 798–802. [Google Scholar] [CrossRef]
- Ma, Z.; Ge, L.; Lee, A.S.Y.; Yong, J.W.H.; Tan, S.N.; Ong, E.S. Simultaneous analysis of different classes of phytohormones in coconut (Cocos nucifera L.) water using high-performance liquid chromatography and liquid chromatography–tandem mass spectrometry after solid-phase extraction. Anal. Chim. Acta 2008, 610, 274–281. [Google Scholar] [CrossRef]
- Weckwerth, W.; Kahl, G. (Eds.) The Handbook of Plant Metabolomics: Metabolite Profiling and Networking, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Aliferis, K.A.; Faubert, D.; Jabaji, S.A. Metabolic Profiling Strategy for the Dissection of Plant Defense against Fungal Pathogens. PLoS ONE 2014, 9, e111930. [Google Scholar] [CrossRef]
- Kachlicki, P.; Piasecka, A.; Stobiecki, M.; Marczak, Ł. Structural characterization of flavonoid glycoconjugates and their derivatives with mass spectrometric techniques. Molecules 2016, 21, 1494. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef]
- Gabelica, V.; Matklund, E. Fundamentals of ion mobility spectrometry. Curr. Opin. Chem. Biol. 2018, 43, 51–59. [Google Scholar] [CrossRef]
- Wiseman, J.M.; Ifa, D.R.; Song, Q.; Cooks, R.G. Tissue imaging at atmospheric pressure using desorption electrospray ionization (DESI) mass spectrometry. Angew. Chem. Int. Ed. 2006, 45, 7188–7192. [Google Scholar] [CrossRef]
- Boughton, B.A.; Thinagaran, D.; Sarabia, D.; Bacic, A.; Roessner, U. Mass spectrometry imaging for plant biology: A review. Phytochem. Rev. 2016, 15, 445–488. [Google Scholar] [CrossRef]
- Heyman, H.M.; Dubery, I.A. The potential of mass spectrometry imaging in plant metabolomics: A review. Phytochem. Rev. 2016, 15, 297–316. [Google Scholar] [CrossRef]
- Doheny-Adams, T.; Redeker, K.; Kittipol, V.; Bancroft, I.; Hartley, S.E. Development of an efficient glucosinolate extraction method. Plant Methods 2017, 13, 17. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Metabolomics in Plant Stress Physiology. In Plant Genetics and Molecular Biology. Advances in Biochemical Engineering/Biotechnology; Varshney, R., Pandey, M., Chitikineni, A., Eds.; Springer: Cham, Switzerland, 2018; p. 164. [Google Scholar]
- Chokkathukalam, A.; Kim, D.H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef]
- Onda, Y.; Hashimoto, K.; Yoshida, T.; Sakurai, T.; Masami, S.; Hirai, Y.; Toyooka, K.; Mochida, K.; Shinozaki, K. Determination of growth stages and metabolic profiles in Brachypodium distachyon for comparison of developmental context with Triticeae crops. Proc. R. Soc. B 2015, 282, 20150964. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef]
- Gorrochategui, E.; Jaumot, J.; Tauler, T. A protocol for LC-MS metabolomic data processing using chemometric tools. Protoc. Exch. 2015. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Ibarra, A.A.G.; Wrobel, K.; Barrientos, E.Y.; Corrales Escobosa, A.R.; Gutierrez Corona, J.F.; Enciso Donis, I.; Wrobel, K. Changes of Metabolomic Profile in Helianthus annuus under Exposure to Chromium (VI) Studied by capHPLC-ESI-QTOF-MS and MS/MS. J. Anal. Methods Chem. 2017, 2017, 3568621. [Google Scholar]
- Mhlongo, M.I.; Steenkamp, P.A.; Piater, L.A.; Madala, N.E.; Dubery, I.A. Profiling of Altered Metabolomic States in Nicotiana tabacum Cells Induced by Priming Agents. Front. Plant Sci. 2016, 7, 1527. [Google Scholar] [CrossRef]
- Montenegro-Burke, J.R.; Aisporna, A.E.; Benton, H.P.; Rinehart, D.; Fang, M.; Huan, T.; Warth, B.; Forsberg, B.; Abe, B.T.; Wolan, D.W.; et al. Data Streaming for Metabolomics: Accelerating Data Processing and Analysis from Days to Minutes. Anal. Chem. 2016, 89, 1254–1259. [Google Scholar] [CrossRef]
- Kessler, N.; Neuweger, H.; Bonte, A.; Langenkämper, G.; Niehaus, K.; Nattkemper, T.W.; Goesmann, A. MeltDB 2.0—Advances of the metabolomics software system. Bioinformatics 2013, 1, 2452–2459. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef]
- Sakurai, T.; Yamada, Y.; Sawada, Y.; Matsuda, F.; Akiyama, K.; Shinozaki, K.; Hirai, M.Y.; Saito, K. PRIMe Update: Innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 2013, 54, e5. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2014, 46, W486–W494. [Google Scholar] [CrossRef]
- Wanichthanarak, K.; Fan, S.; Grapov, D.; Barupal, D.K.; Fiehn, O. Metabox: A Toolbox for Metabolomic Data Analysis, Interpretation and Integrative Exploration. PLoS ONE 2017, 12, e0171046. [Google Scholar] [CrossRef]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Lommen, A.; Kools, H.J. MetAlign 3.0: Performance enhancement by efficient use of advances in computer hardware. Metabolomics 2011, 8, 719–726. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, K.; VanderGheynst, J.; Fiehn, O.; Arita, M.; et al. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Kiefer, P.; Schmitt, U.; Vorholt, J.A. eMZed: An open source framework in Python for rapid and interactive development of LC/MS data analysis workflows. Bioinformatics 2013, 29, 963–964. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Jankevics, A.; Jansen, R.C.; Swertz, M.A.; Breitling, R. PeakML/mzMatch: A File Format, Java Library, R Library, and Tool-Chain for Mass Spectrometry Data Analysis. Anal. Chem. 2011, 83, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Jankevics, A.; Burgess, K.E.V.; Breitling, R.; Barrett, M.P. IDEOM: An Excel interface for analysis of LC–MS-based metabolomics data. Bioinformatics 2012, 28, 1048–1049. [Google Scholar] [CrossRef]
- Zhang, W.; Chang, J.; Lei, Z.; Huhman, D.; Sumner, L.W.; Zhao, P.X. MET-COFEA: A Liquid Chromatography/Mass Spectrometry Data Processing Platform for Metabolite Compound Feature Extraction and Annotation. Anal. Chem. 2014, 86, 6245–6253. [Google Scholar] [CrossRef]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 14, 14.11.1–14.11.23. [Google Scholar]
- Chang, H.Y.; Chen, C.T.; Lih, T.M.; Lynn, K.S.; Juo, C.G.; Hsu, W.L.; Sung, T.Y. iMet-Q: A User-Friendly Tool for Label-Free Metabolomics Quantitation Using Dynamic Peak-Width Determination. PLoS ONE 2016, 11, e0146112. [Google Scholar] [CrossRef] [PubMed]
- Kaever, A.; Landesfeind, M.; Feussner, K.; Mosblech, A.; Heilmann, I.; Morgenstern, B.; Feussner, I.; Meinicke, P. MarVis-Pathway: Integrative and exploratory pathway analysis of non-targeted metabolomics data. Metabolomics 2014, 11, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Tengstrand, E.; Lindberg, J.; Åberg, K.M. TracMass 2: A modular suite of tools for processing chromatography-full scan mass spectrometry data. Anal. Chem. 2014, 86, 3435–3442. [Google Scholar] [CrossRef]
- Hamzeiy, H.; Cox, J. What computational non-targeted mass spectrometry-based metabolomics can gain from shotgun proteomics. Curr. Opin. Biotechnol. 2017, 43, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Pfeuffer, T.; Sachsenber, O.; Alka, M.; Walzer, A.; Fillbrunn, A.; Nilse, L.; Schilling, O.; Reiner, K.; Kohlbacher, O. OpenMS—A platform for reproducible analysis of mass spectrometry data. J. Biotechnol. 2017, 261, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, V.; Hoehenwarter, W.; Lyon, D.; Weckwerth, W.; Wienkoop, S. Using ProtMAX to create high-mass-accuracy precursor alignments from label-free quantitative mass spectrometry data generated in shotgun proteomics experiments. Nat. Protoc. 2013, 8, 595–601. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Perez de Souza, L.; Naake, T.; Tohge, T.; Fernie, A.R. From chromatogram to analyte to metabolite. How to pick horses for courses from the massive web resources for mass spectral plant metabolomics. Gigascience 2017, 6, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kenar, E.; Franken, H.; Forcisi, S.; Wörmann, K.; Häring, H.-U.; Lehmann, R.; Schmitt-Kopplin, P.; Zell, A.; Kohlbacher, O. Automated Label-free Quantification of Metabolites from Liquid Chromatography–Mass Spectrometry Data. Mol. Cell. Proteom. 2014, 13, 348–359. [Google Scholar] [CrossRef]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amode, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Hoff, K.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treviño, V.; Yañez-Garza, I.L.; Rodriguez-López, C.E.; Urrea-López, R.; Garza-Rodriguez, M.L.; Barrera-Saldaña, H.A.; Tamez-Peña, J.G.; Winkler, R.; Díaz de-la-Garza, R.I. GridMass: A fast two-dimensional feature detection method for LC/MS. J. Mass Spectrom. 2015, 50, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, C.E.; Hernández-Brenes, C.; Treviño, V. Avocado fruit maturation and ripening: Dynamics of aliphatic acetogenins and lipidomic profiles from mesocarp, idioblasts and seed. BMC Plant Biol. 2017, 17, 159. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Rajniak, J.; Barco, B.; Clay, N.K.; Sattely, E.S. A new cyanogenic metabolite in Arabidopsis required for inducible pathogen defence. Nature 2015, 525, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Koutouan, C.; Clerc, V.L.; Baltenweck, R.; Claudel, P.; Halter, D.; Hugueney, P.; Hamama, L.; Suel, A.; Huet, S.; Briard, M.; et al. Link between carrot leaf secondary metabolites and resistance to Alternaria dauci. Sci. Rep. 2018, 8, 13746. [Google Scholar] [CrossRef]
- Berini, J.L.; Brockman, S.A.; Hegeman, A.D.; Reich, P.B.; Muthukrishnan, R.; Montgomery, R.A.; Forester, J.D. Combinations of Abiotic Factors Differentially Alter Production of Plant Secondary Metabolites in Five Woody Plant Species in the Boreal-Temperate Transition Zone. Front. Plant Sci. 2018, 9, 1257. [Google Scholar] [CrossRef]
- Mahieu, N.G.; Spalding, J.L.; Patti, G.J. Warpgroup: Increased precision of metabolomic data processing by consensus integration bound analysis. Bioinformatics 2016, 32, 268–275. [Google Scholar] [CrossRef]
- Du, P.; Kibbe, W.A.; Lin, S.M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065. [Google Scholar] [CrossRef] [Green Version]
- Gifford, I.; Battenberg, K.; Vaniya, A.; Wilson, A.; Tian, L.; Fiehn, O.; Berry, A.M. Distinctive Patterns of Flavonoid Biosynthesis in Roots and Nodules of Datisca glomerata and Medicago spp. Revealed by Metabolomic and Gene Expression Profiles. Front. Plant Sci. 2018, 9, 1463. [Google Scholar] [CrossRef]
- Sarabia, L.D.; Boughton, B.A.; Rupasinghe, T.; van de Meene, A.M.L.; Callahan, D.L.; Hill, C.B.; Roessner, U. High-mass-resolution MALDI mass spectrometry imaging reveals detailed spatial distribution of metabolites and lipids in roots of barley seedlings in response to salinity stress. Metabolomics 2018, 14, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libiseller, G.; Dvorzak, M.; Kleb, U.; Gander, E.; Eisenberg, T.; Madeo, F.; Neumann, S.; Trausinger, G.; Sinner, F.; Pieber, T.; et al. IPO: A tool for automated optimization of XCMS parameters. BMC Bioinform. 2015, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Treutler, H.; Neumann, S. Prediction, Detection, and Validation of Isotope Clusters in Mass Spectrometry Data. Metabolites 2016, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Sambles, C.M.; Salmon, D.L.; Florance, H.; Howard, T.P.; Smirnoff, N.; Nielsen, L.R.; McKinney, L.V.; Kjær, E.D.; Buggs, R.J.A.; Grant, M.; et al. Ash leaf metabolomes reveal differences between trees tolerant and susceptible to ash dieback disease. Sci. Data 2017, 4, 170190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riewe, D.; Wiebach, J.; Altmann, T. Structure Annotation and Quantification of Wheat Seed Oxidized Lipids by High-Resolution LC-MS/MS. Plant Physiol. 2017, 175, 600–618. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Saito, K. Ultrahigh resolution metabolomics for S-containing metabolites. Curr. Opin. Biotechnol. 2017, 43, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheltema, S.; Decuypere, J.C.; Dujardin, D.G.; Watson, R.C.; Jansen, R.C.; Breitling, R. Simple data-reduction method for high-resolution LC–MS data in metabolomics. Bioanalysis 2009, 1, 1551–1557. [Google Scholar] [CrossRef]
- Tomasi, G.; van den Berg, F.; Andersson, C. Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data. J. Chemom. 2004, 18, 231–241. [Google Scholar] [CrossRef]
- Ramaker, H.-J.; Van Sprang, E.; Westerhuis, J.A.; Smilde, A.K. Dynamic time warping of spectroscopic BATCH data. Anal. Chim. Acta 2003, 498, 133–153. [Google Scholar] [CrossRef]
- Prince, J.T.; Marcotte, E.M. Chromatographic Alignment of ESI-LC-MS Proteomics Data Sets by Ordered Bijective Interpolated Warping. Anal. Chem. 2006, 78, 6140–6152. [Google Scholar] [CrossRef]
- Escandón, M.; Meijón, M.; Valledor, L.; Pascual, J.; Pinto, G.; Cañal, M.J. Metabolome Integrated Analysis of High-Temperature Response in Pinus radiata. Front. Plant Sci. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Goufo, P.; Moutinho-Pereira, J.M.; Jorge, T.F.; Correia, C.M.; Oliveira, M.R.; Rosa, E.A.S.; António, C.; Trindade, H. Cowpea (Vigna unguiculata L. Walp.) Metabolomics: Osmoprotection as a Physiological Strategy for Drought Stress Resistance and Improved Yield. Front. Plant Sci. 2017, 8, 586. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ren, W.; Kong, H.; Zhao, C.; Zhao, X.; Lin, X.; Lu, X.; Xu, G. An alignment algorithm for LC-MS-based metabolomics dataset assisted by MS/MS, information. Anal. Chim. Acta 2017, 990, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, Z.; Huhman, D.; Sumner, L.W.; Zhao, P.X. MET-XAlign: A Metabolite Cross-Alignment Tool for LC/MS-Based Comparative Metabolomics. Anal. Chem. 2015, 87, 9114–9119. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Kang, J.; Yu, T. Missing value imputation for LC-MS metabolomics data by incorporating metabolic network and adduct ion relations. Bioinformatics 2018, 34, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Bartel, J.; Krumsiek, J.; Theis, F.J. Statistical methods for the analysis of high-throughput metabolomics data. Comput. Struct. Biotechnol. J. 2013, 4, e201301009. [Google Scholar] [CrossRef] [PubMed]
- Gorrochategui, E.; Jaumot, J.; Lacorte, S.; Tauler, R. Data analysis strategies for targeted and untargeted LC-MS metabolomic studies: Overview and workflow. Trends Anal. Chem. 2016, 82, 425–442. [Google Scholar] [CrossRef]
- Piasecka, A.; Sawikowska, A.; Krajewski, P.; Kachlicki, P. Combined mass spectrometric and chromatographic methods for in-depth analysis of phenolic secondary metabolites in barley leaves. J. Mass Spectrom. 2015, 50, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Wojakowska, A.; Piasecka, A.; García-López, P.M.; Zamora-Natera, F.; Krajewski, P.; Marczak, Ł.; Kachlicki, P.; Stobiecki, M. Structural analysis and profiling of phenolic secondary metabolites of Mexican lupine species using LC–MS technique. Phytochemistry 2013, 92, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of Metabolomic Data Using Support Vector Machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.C.; Hour, A.L.; Liu, L.D. Microarray meta-analysis to explore abiotic stress-specific gene expression patterns in Arabidopsis. Bot. Stud. 2017, 58, 22. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Cortes, D.; Miller, G.; Mittler, R. Metabolomics for plant stress response. Physiol. Plant. 2008, 132, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, K.; Kurihara, Y.; Seki, M.; Shinozaki, K. ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr. Opin. Chem. Biol. 2010, 13, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Caldana, C.; Degenkolbe, T.; Cuadros-Inostroza, A.; Klie, S.; Sulpice, R.; Leisse, A.; Steinhauser, D.; Fernie, A.R.; Willmitzer, L.; Hannah, M.A. High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 2011, 67, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Urano, K.; Yoshiwara, K.; Morishita, Y.; Sakurai, N.; Suzuki, H.; Kojima, M.; Sakakibara, H.; Shibata, D.; Saito, K. Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 2014, 164, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegnér, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014, 8 (Suppl. 2), I1. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Instrumental Method | Selectivity * | Sensitivity | Quantitative Analysis | Drawbacks | Additional Comments |
|---|---|---|---|---|---|
| NMR | Good | Low | Good | Low number of identified compounds | Lack of tools for bioinformatic analysis |
| LC/NMR | High | Low | Good | High cost of analyses, low separation of LC column | Absolute structural characterization possible |
| Direct infusion MS | Low | Good | Acceptable | Ionization competition between compounds | Possible estimation of elemental composition of protonated molecules with high resolution mass analyzers |
| GC/MS a or GC/GC/MS a | Good | High | Acceptable | Need of derivatization, low molecular mass range only up to 500 Da | Good separation of compounds with GC column, improved with the use of the GC/GC technique |
| GC/MS/MS a | Good | High | Good | As above | As above |
| LC/MS a | Good | High | Acceptable | Low separation of LC column | Possible estimation of elemental composition of protonated molecules with high resolution mass analyzers |
| LC/MS/MS a | Good | High | Good | Low separation of LC column | Possible estimation of elemental composition of protonated molecules with high resolution mass analyzers, possible differentiation of isomeric and isobaric compounds |
| CE/MS a | Good | Very high | Acceptable | Difficulties of stable hyphenation of CE instrument with mass spectrometer | Good separation of compounds with CE instruments |
| Tool | Data Processing | Data Post-Processing | Statistical Analysis | Integration With Other Omics | Annotation to Metabolomics Databases | Annotation to Pathways Databases | References |
|---|---|---|---|---|---|---|---|
| online services | |||||||
| XCMS online | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [66] https://xcmsonline.scripps.edu |
| PRIMe | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [72] http://prime.psc.riken.jp/ |
| MeltDB | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [70] https://meltdb.cebitec.uni-bielefeld.de |
| Workflow4Metabolomics (W4M) | ✓ | ✓ | ✓ | ✓ | ✓ | [71] http://workflow4metabolomics.org | |
| MetaboAnalyst | ✓ | ✓ | ✓ | ✓ | ✓ | [73] https://www.metaboanalyst.ca/ | |
| Metabox | ✓ | ✓ | ✓ | ✓ | ✓ | [74] | |
| MetFrag | ✓ | ✓ | [75] http://c-ruttkies.github.io/MetFrag | ||||
| local installation | |||||||
| MZmine2 | ✓ | ✓ | ✓ | ✓ | ✓ | [76] | |
| MetAlign | ✓ | ✓ | ✓ | ✓ | [77] | ||
| MS-Dial | ✓ | ✓ | ✓ | [78] | |||
| eMZed | ✓ | ✓ | ✓ | ✓ | [79] | ||
| MzMatch | ✓ | ✓ | ✓ | ✓ | [80] | ||
| IDEOM | ✓ | ✓ | ✓ | ✓ | [81] | ||
| MET-COFEA | ✓ | ✓ | ✓ | ✓ | [82] | ||
| MAVEN | ✓ | ✓ | ✓ | [83] | |||
| iMet-Q | ✓ | ✓ | ✓ | [84] | |||
| MarVis | ✓ | ✓ | ✓ | ✓ | [85] | ||
| TracMass 2 | ✓ | [86] | |||||
| MaxQuant | ✓ | ✓ | ✓ | ✓ | [87] | ||
| OpenMS | ✓ | ✓ | ✓ | ✓ | [88] | ||
| ProtMAX | ✓ | ✓ | ✓ | ✓ | [89] | ||
| MS-Finder | ✓ | ✓ | [90] | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piasecka, A.; Kachlicki, P.; Stobiecki, M. Analytical Methods for Detection of Plant Metabolomes Changes in Response to Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2019, 20, 379. https://doi.org/10.3390/ijms20020379
Piasecka A, Kachlicki P, Stobiecki M. Analytical Methods for Detection of Plant Metabolomes Changes in Response to Biotic and Abiotic Stresses. International Journal of Molecular Sciences. 2019; 20(2):379. https://doi.org/10.3390/ijms20020379
Chicago/Turabian StylePiasecka, Anna, Piotr Kachlicki, and Maciej Stobiecki. 2019. "Analytical Methods for Detection of Plant Metabolomes Changes in Response to Biotic and Abiotic Stresses" International Journal of Molecular Sciences 20, no. 2: 379. https://doi.org/10.3390/ijms20020379
APA StylePiasecka, A., Kachlicki, P., & Stobiecki, M. (2019). Analytical Methods for Detection of Plant Metabolomes Changes in Response to Biotic and Abiotic Stresses. International Journal of Molecular Sciences, 20(2), 379. https://doi.org/10.3390/ijms20020379