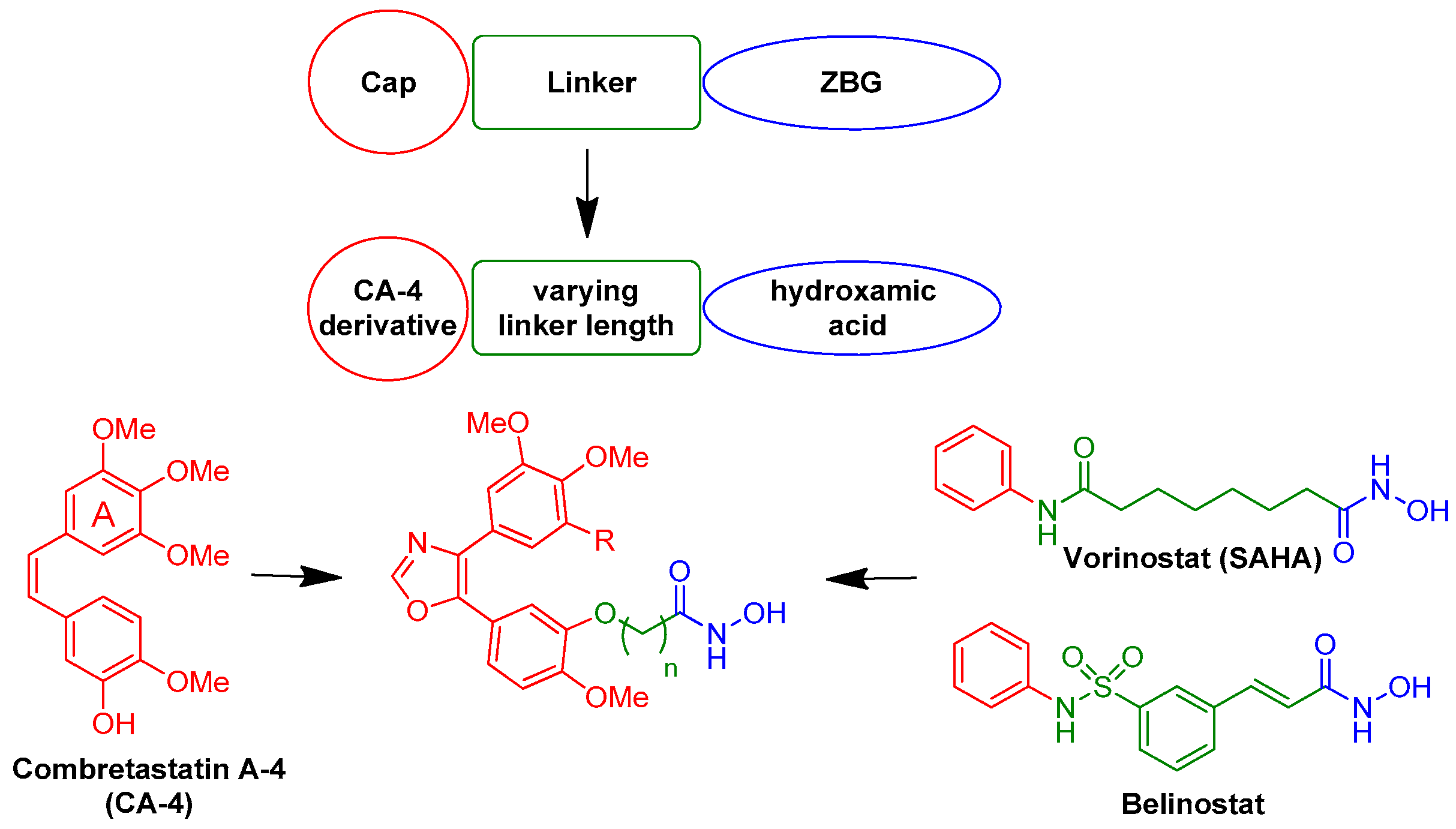
4. Materials and Methods
4.1. General Procedures
The following instruments were used: melting points (uncorrected), Gallenkamp; IR spectra, Perkin-Elmer Spectrum One FT-IR spectrophotometer with ATR sampling unit; nuclear magnetic resonance spectra, BRUKER Avance 300 spectrometer; chemical shifts are given in parts per million (δ) downfield from tetramethylsilane as internal standard; mass spectra, Varian MAT 311A (EI) or UPLC/Orbitrap MS system (ESI); microanalyses, Perkin-Elmer 2400 CHN elemental analyzer. All tested compounds were >95% pure by elemental analysis.
4.2. Materials
The known starting compounds
1a and
2a–
c were prepared according to literature procedures [
25,
26,
27]. The new compounds
1b and
1c were synthesized analogously to
1a (see below). All other starting compounds and reagents were purchased from Sigma–Aldrich.
4.3. General Procedure for the Synthesis of Intermediates 1b and 1c
Isovanillin (152 mg, 1.0 mmol) was dissolved in MeCN (5 mL) and cesium carbonate (652 mg, 2.0 mmol) was added. After stirring at 90 °C for 0.5 h, ethyl 5-bromovalerate (328 µL, 2.0 mmol) or ethyl 6-bromohexanoate (356 µL, 2.0 mmol), respectively, was added and the reaction mixture was stirred at 90 °C for 2 h. After filtration through celite, the solvent was removed under reduced pressure and the residue was washed twice with n-hexane. The resulting oil was dried in vacuum and used for the next step without further purification.
Ethyl 5-(1-methoxy-4-formyl-2-phenoxy)valerate (1b). Yield: 130 mg (0.46 mmol, 46%); colorless oil. υmax(ATR)/cm−1 2941, 2881, 2841, 1728, 1683, 1585, 1509, 1462, 1436, 1393, 1374, 1340, 1262, 1238, 1159, 1133, 1096, 1019, 935, 864, 809, 780, 749, 737, 641. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.8–2.0 (m, 4H), 2.38 (t, 2H, J = 7.3 Hz), 3.93 (s, 3H), 4.0–4.2 (m, 4H), 6.95 (d, 1H, J = 8.2 Hz), 7.37 (s, 1H), 7.44 (d, 2H, J = 8.2 Hz), 9.82 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 21.6, 28.5, 33.9, 56.2, 60.3, 68.5, 110.5, 110.7, 126.7, 130.1, 149.0, 154.9, 173.3, 190.9 ppm. m/z (%) 280 (14) [M+], 235 (24), 151 (57), 129 (98), 101 (100), 83 (82), 55 (56), 43 (15).
Ethyl 6-(1-methoxy-4-formyl-2-phenoxy)hexanoate (1c). Yield: 110 mg (0.37 mmol, 37%); colorless oil. υmax(ATR)/cm−1 2940, 2870, 1729, 1685, 1585, 1510, 1462, 1436, 1394, 1374, 1341, 1264, 1239, 1161, 1134, 1068, 1021, 862, 811, 779, 749, 729, 641. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.4–1.6 (m, 2H), 1.6–1.8 (m, 2H), 1.8–1.9 (m, 2H), 2.32 (t, 2H, J = 7.3 Hz), 4.01 (s, 3H), 4.0–4.2 (m, 4H), 6.95 (d, 1H, J = 8.2 Hz), 7.37 (s, 1H), 7.44 (d, 2H, J = 8.2 Hz), 9.82 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.7, 25.6, 28.7, 34.2, 56.2, 60.2, 68.8, 110.4, 110.6, 118.0, 126.6, 130.1, 149.1, 154.9, 173.6, 190.9 ppm. m/z (%) 294 (48) [M+], 249 (35), 152 (98), 143 (100), 115 (54), 97 (97), 69 (86), 55 (22), 41 (28).
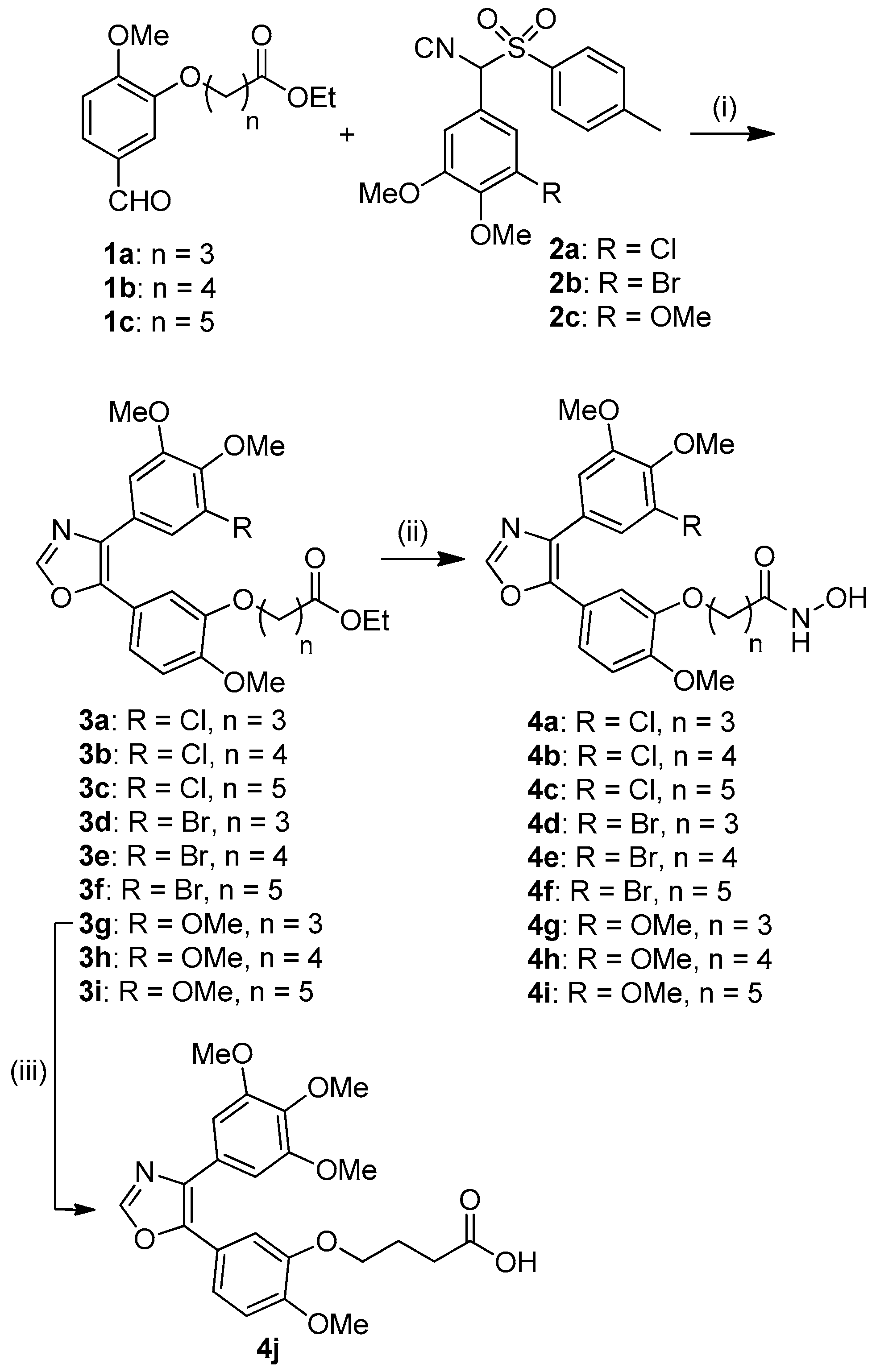
4.4. General Procedure for the Synthesis of Intermediates 3a–i
The respective compounds 1 (0.42 mmol) and 2 (0.42 mmol) and K2CO3 (590 mg, 4.3 mmol) were suspended in ethanol and stirred under reflux for 2 h. The solvent was removed and the residue dissolved in ethyl acetate and washed with water. The organic phase was dried over Na2SO4, concentrated in vacuum, and the residue was purified by column chromatography (silica gel 60).
Ethyl 4-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy] butyrate (3a). Compound 3a was obtained from 1a (113 mg, 0.42 mmol), 2a (155 mg, 0.42 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 103 mg (0.22 mmol, 52%); Rf = 0.39 (ethyl acetate/n-hexane, 2:3); colorless oil. υmax(ATR)/cm−1 2969, 2938, 2876, 2837, 1731, 1683, 1629, 1561, 1513, 1489, 1463, 1443, 1414, 1399, 1366, 1303, 1254, 1229, 1174, 1140, 1117, 1105, 1047, 1024, 1000, 960, 938, 892, 853, 810, 774, 758, 734, 709, 659, 629. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 2.0–2.2 (m, 2H), 2.49 (t, 2H, J = 7.3 Hz), 3.81 (s, 3H), 3.87 (s, 3H), 3.88 (s, 3H), 3.98 (t, 2H, J = 6.3 Hz), 4.11 (q, 2H, J = 7.2 Hz), 6.87 (d, 1H, J = 8.4 Hz), 7.1–7.2 (m, 3H), 7.29 (s, 1H), 7.88 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.5, 30.7, 56.0, 56.1, 60.4, 60.8, 68.1, 110.6, 111.7, 112.0, 120.4, 120.9, 121.1, 128.3, 128.7, 132.3, 146.1, 148.4, 149.3, 150.4, 153.8, 173.0 ppm. m/z (%) 477 (61) [M+], 475 (100) [M+], 432 (11), 430 (31), 361 (8), 265 (7), 115 (94), 87 (67), 43 (11).
Ethyl 5-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]valerate (3b). Compound 3b was obtained from 1b (126 mg, 0.45 mmol), 2a (165 mg, 0.45 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 98 mg (0.20 mmol, 44%); Rf = 0.42 (ethyl acetate/n-hexane, 2:3); colorless oil. υmax(ATR)/cm−1 2938, 2870, 2838, 1730, 1603, 1591, 1564, 1513, 1489, 1463, 1414, 1365, 1324, 1294, 1254, 1229, 1203, 1164, 1140, 1105, 1048, 1024, 938, 854, 811, 776, 758, 710, 659, 628. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.7–1.9 (m, 2H), 2.35 (t, 2H, J = 7.3 Hz), 3.81 (s, 3H), 3.87 (s, 3H), 3.88 (s, 3H), 3.93 (t, 2H, J = 6.1 Hz), 4.10 (q, 2H, J = 7.1 Hz), 6.87 (d, 1H, J = 8.4 Hz), 7.10 (s, 1H), 7.1–7.2 (m, 2H), 7.30 (s, 1H), 7.88 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 21.6, 28.6, 33.9, 56.0, 56.1, 60.3, 60.8, 68.7, 110.6, 111.7, 120.2, 120.9, 121.1, 128.3, 128.7, 132.3, 145.2, 146.1, 148.5, 149.3, 150.3, 153.8, 173.4 ppm. m/z (%) 491 (43) [M+], 489 (100) [M+], 446 (13), 444 (41), 361 (31), 346 (17), 168 (14), 129 (98), 101 (98), 83 (56), 55 (39), 43 (11).
Ethyl 6-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy] hexanoate (3c). Compound 3c was obtained from 1c (91 mg, 0.31 mmol), 2a (113 mg, 0.31 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 63 mg (0.13 mmol, 42%); Rf = 0.45 (ethyl acetate/n-hexane, 2:3); colorless oil. υmax(ATR)/cm−1 2939, 2871, 2837, 1730, 1605, 1591, 1563, 1513, 1489, 1463, 1414, 1366, 1325, 1300, 1255, 1229, 1203, 1175, 1165, 1140, 1105, 1048, 1024, 1001, 939, 854, 821, 775, 758, 710, 659, 629. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.4-1.5 (m, 2H), 1.6–1.7 (m, 2H), 1.8–1.9 (m, 2H), 2.30 (t, 2H, J = 7.3 Hz), 3.80 (s, 3H), 3.87 (s, 3H), 3.88 (s, 3H), 3.92 (t, 2H, J = 6.7 Hz), 4.10 (q, 2H, J = 7.1 Hz), 6.88 (d, 1H, J = 8.4 Hz), 7.10 (s, 1H), 7.1-7.2 (m, 2H), 7.30 (s, 1H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.7, 25.6, 28.8, 34.2, 56.0, 56.1, 60.2, 60.8, 68.9, 110.6, 111.6, 120.1, 120.9, 121.1, 128.3, 128.7, 132.3, 145.2, 146.1, 148.6, 149.3, 150.3, 153.8, 173.6 ppm. m/z (%) 505 (68) [M+], 503 (100) [M+], 458 (11), 361 (37), 346 (13), 143 (51), 115 (16), 97 (28), 69 (26).
Ethyl 4-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy] butyrate (3d). Compound 3d was obtained from 1a (125 mg, 0.47 mmol), 2b (192 mg, 0.47 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 100 mg (0.19 mmol, 41%); Rf = 0.25 (ethyl acetate/n-hexane, 1:2); colorless oil. υmax(ATR)/cm−1 3118, 2986, 2940, 2837, 1739, 1592, 1561, 1514, 1488, 1468, 1441, 1404, 1388, 1362, 1349, 1267, 1256, 1241, 1226, 1208, 1179, 1145, 1124, 1108, 1072, 1048, 1025, 997, 960, 944, 859, 846, 820, 807, 773, 754, 699, 654. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.2 Hz), 2.0–2.2 (m, 2H), 2.50 (t, 2H, J = 7.3 Hz), 3.80 (s, 3H), 3.86 (s, 3H), 3.88 (s, 3H), 3.98 (t, 2H, J = 6.3 Hz), 4.11 (q, 2H, J = 7.2 Hz), 6.88 (d, 1H, J = 8.4 Hz), 7.12 (s, 1H), 7.1–7.2 (m, 2H), 7.46 (s, 1H), 7.88 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.5, 30.8, 56.0, 56.1, 60.4, 60.7, 68.2, 111.4, 111.7, 111.9, 117.6, 120.3, 120.9, 123.9, 129.3, 146.1, 148.4, 149.3, 150.4, 153.7, 173.0 ppm. m/z (%) 521 (38) [M+], 519 (37) [M+], 476 (6), 474 (5), 115 (100), 87 (79), 43 (14).
Ethyl 5-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]valerate (3e). Compound 3e was obtained from 1b (112 mg, 0.40 mmol) and 2b (164 mg, 0.40 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 110 mg (0.21 mmol, 53%); Rf = 0.33 (ethyl acetate/n-hexane, 2:3); colorless oil. υmax(ATR)/cm−1 3128, 2938, 2838, 1730, 1591, 1557, 1513, 1485, 1463, 1414, 1364, 1254, 1230, 1201, 1174, 1162, 1140, 1105, 1043, 1025, 1000, 939, 897, 854, 807, 776, 754, 698, 659, 628. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.7–1.9 (m, 4H), 2.36 (t, 2H, J = 7.3 Hz), 3.80 (s, 3H), 3.86 (s, 3H), 3.88 (s, 3H), 3.93 (t, 2H, J = 6.1 Hz), 4.10 (q, 2H, J = 7.1 Hz), 6.88 (d, 1H, J = 8.4 Hz), 7.11 (s, 1H), 7.1–7.2 (m, 2H), 7.47 (s, 1H), 7.88 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 21.6, 28.6, 33.9, 56.0, 56.1, 60.3, 60.7, 68.7, 111.4, 111.6, 111.7, 117.6, 120.1, 120.9, 123.9, 129.4, 132.1, 146.1, 146.3, 148.6, 149.3, 150.3, 153.7, 173.3 ppm. m/z (%) 535 (95) [M+], 533 (95) [M+], 490 (17), 488 (16), 407 (15), 405 (15), 129 (96), 101 (100), 83 (42), 55 (40).
Ethyl 6-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy] hexanoate (3f). Compound 3f was obtained from 1c (113 mg, 0.38 mmol), 2b (158 mg, 0.38 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 80 mg (0.15 mmol, 40%); Rf = 0.45 (ethyl acetate/n-hexane, 2:3); colorless oil. υmax(ATR)/cm−1 2940, 2869, 1730, 1590, 1558, 1513, 1485, 1463, 1414, 1364, 1323, 1253, 1229, 1175, 1160, 1140, 1105, 1066, 1043, 1025, 999, 938, 853, 806, 775, 753, 697, 659, 628. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.4–1.5 (m, 2H), 1.6–1.8 (m, 2H), 1.8–1.9 (m, 2H), 2.30 (t, 2H, J = 7.3 Hz), 3.80 (s, 3H), 3.86 (s, 3H), 3.88 (s, 3H), 3.9-4.0 (m, 5H), 4.10 (q, 2H, J = 7.1 Hz), 6.88 (d, 1H, J = 8.4 Hz), 7.10 (s, 1H), 7.1–7.2 (m, 2H), 7.47 (s, 1H), 7.88 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.7, 25.6, 28.8, 34.2, 56.0, 56.1, 60.2, 60.7, 68.9, 111.4, 111.6, 111.7, 117.6, 120.0, 120.9, 123.9, 129.4, 132.1, 146.2, 148.6, 149.3, 150.3, 153.7, 173.6 ppm. m/z (%) 549 (99) [M+], 547 (100) [M+], 407 (31), 405 (32), 143 (77), 115 (20), 97 (32), 69 (27).
Ethyl 4-[1-methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]butyrate (3g). Compound 3g was obtained from 1a (134 mg, 0.50 mmol), 2c (182 mg, 0.50 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 124 mg (0.26 mmol, 52%); Rf = 0.31 (ethyl acetate/n-hexane, 1:1); colorless oil. υmax(ATR)/cm−1 2939, 2840, 1730, 1583, 1515, 1463, 1415, 1372, 1306, 1255, 1236, 1173, 1122, 1022, 1004, 956, 938, 885, 837, 811, 768, 733, 659, 629. 1H NMR (300 MHz, CDCl3): δ = 1.27 (t, 3H, J = 7.1 Hz), 2.1–2.2 (m, 2H), 2.53 (t, 2H, J = 7.3 Hz), 3.81 (s, 6H), 3.90 (s, 3H), 3.91 (s, 3H), 4.02 (t, 2H, J = 6.3 Hz), 4.15 (q, 2H, J = 7.1 Hz), 6.90 (d, 1H, J = 8.4 Hz), 7.12 (s, 1H), 6.94 (s, 2H), 7.17 (s, 1H), 7.25 (d, 1H, J = 8.4 Hz), 7.93 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.5, 30.7, 56.0, 56.1, 60.4, 60.9, 68.0, 104.9, 111.5, 112.1, 120.4, 121.3, 127.6, 133.6, 137.9, 145.6, 148.3, 149.2, 150.1, 153.3, 173.0 ppm. m/z (%) 471 (95) [M+], 456 (13), 426 (31), 195 (17), 115 (100), 87 (99), 69 (14), 43 (24). Elemental analysis calculated (%) for C25H29NO8: C 63.68, H 6.20, N 2.97. Found: C 63.65, H 6.18, N 2.96.
Ethyl 5-[1-methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]valerate (3h). Compound 3h was obtained from 1b (130 mg, 0.46 mmol), 2c (168 mg, 0.46 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 107 mg (0.22 mmol, 48%); Rf = 0.35 (ethyl acetate/n-hexane, 1:1); colorless oil. υmax(ATR)/cm−1 2938, 2840, 1730, 1583, 1515, 1455, 1415, 1372, 1330, 1305, 1255, 1237, 1171, 1123, 1022, 1004, 938, 894, 837, 811, 768, 734, 659, 629. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.7–1.9 (m, 4H), 2.34 (d, 2H, J = 7.2 Hz), 3.77 (s, 6H), 3.85 (s, 3H), 3.87 (s, 3H), 3.93 (t, 2H, J = 6.1 Hz), 4.10 (q, 2H, J = 7.1 Hz), 6.86 (d, 1H, J = 8.4 Hz), 6.90 (s, 2H), 7.11 (s, 1H), 7.20 (d, 1H, J = 8.4 Hz), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 21.6, 28.6, 33.9, 56.0, 56.1, 60.3, 60.9, 68.6, 105.0, 111.5, 111.9, 120.3, 121.3, 127.6, 133.6, 138.0, 145.7, 148.5, 149.2, 150.1, 153.3, 173.3 ppm. m/z (%) 485 (100) [M+], 440 (15), 401 (16), 153 (16), 129 (99), 101 (73), 83 (26), 55 (17).
Ethyl 6-[1-methoxy-4-(4’-(3’’,4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]hexanoate (3i). Compound 3i was obtained from 1c (116 mg, 0.39 mmol), 2c (114 mg, 0.46 mmol) and K2CO3 (590 mg, 4.3 mmol). Yield: 86 mg (0.17 mmol, 44%); Rf = 0.45 (ethyl acetate/n-hexane, 1:1); colorless oil. υmax(ATR)/cm−1 2938, 2870, 2839, 1730, 1583, 1515, 1462, 1415, 1372, 1327, 1305, 1255, 1236, 1173, 1123, 1022, 1005, 939, 891, 836, 811, 769, 734, 659, 630. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 3H, J = 7.1 Hz), 1.4–1.5 (m, 2H), 1.6–1.8 (m, 2H), 1.8–1.9 (m, 2H), 2.30 (d, 2H, J = 7.3 Hz), 3.77 (s, 6H), 3.86 (s, 3H), 3.87 (s, 3H), 3.91 (t, 2H, J = 6.6 Hz), 4.10 (q, 2H, J = 7.1 Hz), 6.86 (d, 1H, J = 8.4 Hz), 6.90 (s, 2H), 7.11 (s, 1H), 7.20 (d, 1H, J = 8.4 Hz), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 14.2, 24.7, 25.6, 28.8, 34.2, 56.0, 56.1, 60.2, 60.9, 68.8, 105.0, 111.5, 111.8, 120.2, 121.3, 127.7, 133.6, 138.0, 145.7, 148.5, 149.2, 150.1, 153.3, 173.6 ppm. m/z (%) 499 (100) [M+], 484 (22), 454 (15), 357 (12), 342 (17), 249 (15), 154 (53), 143 (99), 115 (27), 97 (58), 69 (43).
4.5. General Procedure for the Synthesis of Compounds 4a–i
The corresponding compound 3 (0.20 mmol) was dissolved in CH2Cl2/MeOH (9 mL, 1:2), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol) were added and the reaction mixture was stirred at room temperature for 1 h. The solvent was removed, the residue was dissolved in water and adjusted to pH 7–8 with acetic acid. The aqueous phase was extracted with ethyl acetate (2 × 50 mL), dried over Na2SO4 and concentrated in vacuum. The solid residue was recrystallized from CH2Cl2/n-hexane.
N-Hydroxy-4-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]butanamide (4a). Compound 4a was obtained from 3a (97 mg, 0.20 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 64 mg (0.14 mmol, 70%); colorless solid of mp 171–172 °C. υmax(ATR)/cm−1 3101, 2942, 2868, 2843, 1655, 1593, 1566, 1516, 1492, 1463, 1441, 1401, 1364, 1328, 1258, 1242, 1228, 1206, 1179, 1168, 1141, 1117, 1100, 1073, 1052, 1024, 997, 941, 887, 866, 847, 808, 755, 710, 655, 625. 1H NMR (300 MHz, CDCl3): δ = 2.0–2.2 (m, 2H), 2.4–2.5 (m, 2H), 3.82 (s, 3H), 3.88 (s, 3H), 3.88 (s, 3H), 3.9–4.0 (m, 5H), 6.90 (d, 1H, J = 8.5 Hz), 7.07 (s, 1H), 7.15 (s, 1H), 7.2–7.3 (m, 3H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.4, 30.5, 56.0, 56.2, 60.9, 68.4, 110.8, 111.3, 111.5, 120.6, 121.0, 121.2, 128.3, 128.7, 132.4, 145.2, 145.8, 147.8, 149.4, 149.8, 153.9, 170.6 ppm. m/z (%) 464 (4) [M+], 462 (10) [M+], 447 (27), 363 (32), 361 (100), 346 (19), 318 (10), 86 (12). Elemental analysis calculated (%) for C22H23ClN2O7: C 57.09, H 5.01, N 6.05. Found: C 57.06, H 5.02, N 6.04.
N-Hydroxy-5-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]pentanamide (4b). Compound 4b was obtained from 3b (90 mg, 0.18 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 62 mg (0.13 mmol, 72%); colorless solid of mp 202 °C. υmax(ATR)/cm−1 3194, 2942, 2870, 2837, 1644, 1606, 1592, 1564, 1513, 1489, 1460, 1440, 1400, 1364, 1326, 1255, 1230, 1206, 1176, 1140, 1107, 1047, 1019, 996, 939, 895, 853, 807, 759, 711, 657, 628, 607. 1H NMR (300 MHz, CDCl3): δ = 1.7–1.9 (m, 4H), 2.3–2.4 (m, 2H), 3.82 (s, 3H), 3.88 (s, 3H), 3.9–4.0 (m, 5H), 6.91 (d, 1H, J = 8.4 Hz), 7.04 (s, 1H), 7.1-7.3 (m, 3H), 7.90 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 21.5, 24.4, 26.4, 32.1, 55.9, 56.2, 60.9, 69.8, 110.2, 110.8, 111.3, 111.7, 119.9, 121.2, 128.3, 128.8, 132.4, 145.1, 146.0, 148.1, 149.2, 149.4, 153.9, 170.2 ppm. m/z (%) 476 (3) [M+], 461 (42), 363 (33), 361 (100), 346 (24), 318 (16), 240 (10), 100 (41), 44 (56). Elemental analysis calculated (%) for C23H25ClN2O7: C 57.92, H 5.28, N 5.87. Found: C 57.90, H 5.26, N 5.86.
N-Hydroxy-6-[1-methoxy-4-(4’-(3’’-chloro-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]hexanamide (4c). Compound 4c was obtained from 3c (58 mg, 0.12 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 44 mg (0.09 mmol, 75%); colorless solid of mp 189 °C. υmax(ATR)/cm−1 3180, 3120, 2997, 2939, 2865, 2837, 1652, 1595, 1567, 1513, 1491, 1464, 1435, 1399, 1363, 1327, 1305, 1257, 1228, 1206, 1192, 1175, 1141, 1110, 1074, 1049, 1017, 996, 951, 895, 870, 851, 823, 807, 777, 758, 729, 709, 657, 630. 1H NMR (300 MHz, CDCl3): δ = 1.4–1.5 (m, 2H), 1.6–1.7 (m, 2H), 1.7–1.8 (m, 2H), 2.1-2.2 (m, 2H), 3.8-3.9 (m, 11H), 6.90 (d, 1H, J = 8.4 Hz), 7.04 (s, 1H), 7.15 (s, 1H), 7.21 (d, 1H, J = 8.4 Hz), 7.28 (s, 1H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.9, 25.5, 28.3, 32.8, 56.0, 56.2, 60.9, 68.9, 110.8, 111.3, 111.8, 119.9, 120.9, 121.3, 128.3, 129.0, 132.2, 145.0, 146.1, 148.4, 149.4, 150.0, 153.8, 170.9 ppm. m/z (%) 491 (5) [M+], 489 (14) [M+], 477 (23), 475 (72), 363 (23), 361 (100), 348 (13), 346 (36), 318 (23), 182 (13), 114 (45), 69 (51). Elemental analysis calculated (%) for C24H27ClN2O7: C 58.72, H 5.54, N 5.71. Found: C 58.70, H 5.56, N 5.69.
N-Hydroxy-4-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]butanamide (4d). Compound 4d was obtained from 3d (100 mg, 0.19 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 86 mg (0.17 mmol, 90%); colorless solid of mp 140–141 °C. υmax(ATR)/cm−1 3192, 2934, 2837, 1639, 1592, 1559, 1513, 1486, 1463, 1440, 1400, 1362, 1255, 1229, 1179, 1140, 1107, 1076, 1041, 1021, 994, 954, 888, 856, 844, 806, 753, 733, 697, 657, 627. 1H NMR (300 MHz, CDCl3): δ = 2.0–2.2 (m, 2 H), 2.4–2.5 (m, 2H), 3.82 (s, 3H), 3.87 (s, 3H), 3.9–4.0 (m, 5H), 6.91 (d, 1H, J = 8.5 Hz), 7.08 (s, 1H), 7.19 (s, 1H), 7.2–7.3 (m, 1H), 7.43 (s, 1H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.4, 30.6, 56.0, 56.2, 60.7, 68.5, 111.3, 111.6, 117.6, 120.5, 121.1, 124.0, 129.4, 132.3, 145.9, 146.2, 147.8, 149.4, 149.8, 153.8, 170.5 ppm. m/z (%) 508 (7) [M+], 506 (8) [M+], 493 (17), 491 (20), 407 (66), 405 (67), 102 (26), 86 (32), 33 (100). Elemental analysis calculated (%) for C22H23BrN2O7: C 52.08, H 4.57, N 5.52. Found: C 52.06, H 4.56, N 5.50.
N-Hydroxy-5-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]pentanamide (4e). Compound 4e was obtained from 3e (110 mg, 0.21 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 74 mg (0.14 mmol, 67%); colorless solid of mp 152–153 °C. υmax(ATR)/cm−1 3186, 2940, 1641, 1593, 1563, 1515, 1487, 1401, 1361, 1327, 1257, 1231, 1181, 1162, 1143, 1106, 1074, 1040, 1022, 993, 950, 894, 858, 845, 802, 753, 700, 655, 628. 1H NMR (300 MHz, CDCl3): δ = 1.8–1.9 (m, 4 H), 2.3–2.5 (m, 2H), 3.82 (s, 3H), 3.87 (s, 3H), 3.9–4.0 (m, 5H), 6.90 (d, 1H, J = 8.5 Hz), 7.05 (s, 1H), 7.2–7.3 (m, 2H), 7.44 (s, 1H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.4, 26.4, 32.1, 55.9, 56.2, 60.8, 69.8, 110.1, 111.3, 111.6, 117.6, 119.9, 121.2, 124.0, 129.5, 132.3, 146.0, 146.2, 148.1, 149.2, 149.4, 153.7, 170.3 ppm. m/z (%) 521 (5) [M+], 519 (5) [M+], 505 (24), 407 (100), 405 (95), 392 (24), 390 (24), 364 (12), 362 (13), 100 (93), 72 (67), 55 (54). Elemental analysis calculated (%) for C23H25BrN2O7: C 52.99, H 4.83, N 5.37. Found: C 52.97, H 4.82, N 5.35.
N-Hydroxy-6-[1-methoxy-4-(4’-(3’’-bromo-4’’,5’’-dimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]hexanamide (4f). Compound 4f was obtained from 3f (80 mg, 0.15 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 75 mg (0.14 mmol, 93%); colorless solid of mp 115–117 °C. υmax(ATR)/cm−1 3190, 2938, 2865, 1647, 1591, 1558, 1512, 1485, 1463, 1400, 1362, 1325, 1254, 1229, 1203, 1176, 1139, 1107, 1042, 998, 939, 853, 807, 775, 753, 697, 658, 628. 1H NMR (300 MHz, CDCl3): δ = 1.4–1.5 (m, 2H), 1.6–1.7 (m, 2H), 1.7–1.8 (m, 4H), 2.2–2.3 (m, 2H), 3.80 (s, 3H), 3.8–3.9 (m, 8H), 6.89 (d, 1H, J = 8.5 Hz), 7.05 (s, 1H), 7.2–7.3 (m, 2H), 7.43 (s, 1H), 7.89 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 25.0, 25.5, 28.3, 32.8, 56.0, 56.1, 60.8, 68.9, 111.2, 111.6, 111.7, 117.6, 119.9, 120.9, 124.1, 129.6, 132.0, 146.0, 146.1, 148.4, 149.4, 150.0, 153.7, 171.0 ppm. m/z (%) 536 (1) [M+], 534 (1) [M+], 521 (91), 519 (100), 407 (53), 405 (51), 392 (17), 390 (17), 364 (7), 362 (8), 282 (7), 265 (12), 243 (7), 194 (16), 167 (28), 114 (36), 69 (17), 44 (30). Elemental analysis calculated (%) for C24H27BrN2O7: C 53.84, H 5.08, N 5.23. Found: C 53.82, H 5.06, N 5.22.
N-Hydroxy-4-[1-methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]butanamide (4g). Compound 4g was obtained from 3g (119 mg, 0.25 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 74 mg (0.16 mmol, 64%); colorless solid of mp 121–123°C. υmax(ATR)/cm−1 3194, 2935, 2837, 1658, 1583, 1515, 1454, 1415, 1373, 1256, 1238, 1173, 1123, 1020, 1002, 939, 884, 838, 813, 767, 734, 658, 628. 1H NMR (300 MHz, CDCl3) δ 2.0–2.2 (m, 2H), 2.3–2.4 (m, 2H), 3.76 (s, 6H), 3.85 (s, 3H), 3.9–4.0 (m, 5H), 4.15 (q, 2H, J = 7.1 Hz), 6.8–6.9 (m, 3H), 7.05 (s, 1H), 7.2–7.3 (m, 1H), 7.90 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.5, 30.2, 56.0, 56.2, 61.0, 68.2, 105.2, 110.0, 111.4, 120.4, 121.3, 127.8, 133.7, 145.5, 147.7, 149.3, 149.7, 153.3, 170.5 ppm. m/z (%) 458 (4) [M+], 443 (100), 428 (26), 357 (98), 342 (62), 314 (17), 236 (16), 195 (18), 151 (12), 86 (22), 44 (29). Elemental analysis calculated (%) for C23H26N2O8: C 60.26, H 5.72, N 6.11. Found: C 60.23, H 5.70, N 6.08.
N-Hydroxy-5-[1-methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]pentanamide (4h). Compound 4h was obtained from 3h (98 mg, 0.20 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 72 mg (0.15 mmol, 76%); colorless solid of mp 118–120°C. υmax(ATR)/cm−1 3191, 2937, 2870, 2843, 1657, 1584, 1515, 1454, 1415, 1373, 1306, 1255, 1239, 1173, 1123, 1022, 838, 811, 768, 734, 658, 629. 1H NMR (300 MHz, CDCl3): δ = 1.8–1.9 (m, 4H), 2.2–2.3 (m, 2H), 3.78 (s, 6H), 3.8–3.9 (m, 5H), 3.94 (s, 3H), 6.8-6.9 (m, 3H), 7.02 (s, 1H), 7.2–7.3 (m, 1H), 7.90 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.0, 26.6, 32.0, 55.9, 56.2, 61.0, 69.5, 105.3, 110.4, 111.2, 119.9, 121.5, 127.9, 133.7, 137.8, 145.6, 148.0, 149.1, 149.3, 153.3, 170.1 ppm. m/z (%) 472 (3) [M+], 457 (100), 442 (10), 357 (73), 342 (34), 314 (11), 100 (17), 44 (12). Elemental analysis calculated (%) for C24H28N2O8: C 61.01, H 5.97, N 5.93. Found: C 61.04, H 5.95, N 5.90.
N-Hydroxy-6-[1-methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]hexanamide (4i). Compound 4i was obtained from 3i (73 mg, 0.15 mmol), hydroxylamine (50% in water, 0.5 mL, 15 mmol) and NaOH (200 mg, 5 mmol). Yield: 52 mg (0.11 mmol, 73%); colorless solid of mp 114–116 °C. υmax(ATR)/cm−1 3211, 2936, 2870, 2837, 1651, 1584, 1515, 1455, 1414, 1372, 1331, 1306, 1255, 1238, 1173, 1122, 1019, 1001, 939, 892, 836, 810, 767, 732, 658, 628. 1H NMR (300 MHz, CDCl3): δ = 1.3–1.4 (m, 2H), 1.5–1.7 (m, 4H), 2.1–2.2 (m, 2H), 3.76 (s, 6H), 3.8–3.9 (m, 8H), 6.8-6.9 (m, 3H), 7.03 (s, 1H), 7.2–7.3 (m, 1H), 7.90 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 25.0, 25.4, 28.3, 32.8, 56.0, 56.1, 61.0, 68.8, 105.2, 111.5, 111.6, 119.8, 121.2, 127.9, 133.4, 137.6, 145.7, 148.2, 149.3, 149.8, 153.2, 171.0 ppm. m/z (%) 486 (18) [M+], 471 (100), 456 (17), 443 (11), 357 (48), 342 (38), 193 (17), 114 (13), 44 (21). Elemental analysis calculated (%) for C25H30N2O8: C 61.72, H 6.22, N 5.76. Found: C 61.70, H 6.21, N 5.74.
4.6. Ester Hydrolysis of 3g to Carboxylic Acid 4j
4-[1-Methoxy-4-(4’-(3’’,4’’,5’’-trimethoxyphenyl)oxazol-5’-yl)-2-phenoxy]butyric acid (4j). Compound 3g (119 mg, 0.25 mmol) was dissolved in MeOH (10 mL), aqueous NaOH (1 M, 10 mL) was added and the reaction mixture was stirred at room temperature for 24 h. The solution was acidified with aqueous HCl (1 M, to pH < 2) and the resulting precipitate was collected, washed with water, and dried in vacuum. Yield: 75 mg (0.17 mmol, 68%); colorless solid of mp 209–210 °C. υmax(ATR)/cm−1 3134, 2940, 2873, 2835, 2538, 1728, 1606, 1586, 1519, 1474, 1446, 1414, 1372, 1316, 1274, 1251, 1237, 1196, 1173, 1147, 1128, 1112, 1068, 1043, 1031, 1022, 1007,962, 946, 883, 855, 840, 804, 769, 761, 736, 674, 654, 630, 623. 1H NMR (300 MHz, CDCl3): δ = 2.0–2.2 (m, 2H), 2.56 (t, 2H, J = 7.2 Hz), 3.76 (s, 6H), 3.86 (s, 3H), 3.87 (s, 3H), 3.98 (t, 1H, J = 6.3 Hz), 6.86 (d, 1H, J = 8.5 Hz), 6.88 (s, 2H), 7.12 (s, 1H), 7.22 (d, 1H, J = 8.5 Hz), 7.91 ppm (s, 1H). 13C NMR (75.5 MHz, CDCl3): δ = 24.2, 30.2, 56.0, 56.1, 60.9, 67.8, 105.1, 111.6, 112.1, 120.5, 121.3, 127.6, 133.5, 137.9, 145.6, 148.2, 149.3, 150.1, 153.3, 177.0 ppm. m/z (ESI, %) 466.2 (27) [M+ + Na], 444.2 (100) [M+]. Elemental analysis calculated (%) for C23H25NO8: C 62.30, H 5.68, N 3.16. Found: C 62.26, H 5.65, N 3.08.
4.7. Biological Evaluations
4.7.1. Cell Lines and Culture Conditions
HT-29 (ACC-299), HCT-116 (ACC-581) and DLD-1 (ACC-278) colon carcinoma, MCF-7 (ACC-115) breast carcinoma, KB-V1 (ACC-149), 518A2 (Department of Radiotherapy and Radiobiology, University Hospital Vienna) melanoma and Ea.Hy926 (ATCC no. CRL-2922) HUVEC derived endothelial hybrid cells line were grown in Dulbecco’s Modified Eagle Medium (DMEM; Biochrom), high glucose supplemented with 10% (v/v) fetal bovine serum (FBS; Biochrom.) and 1% (v/v) Antibiotic-Antimycotic solution (anti-anti; Thermo Scientific). HDFa human dermal fibroblasts (ATCC: PCS-201-012™) were grown in DMEM supplemented with 10% FBS, 1% anti-anti and 2 mM glutamine. The cells were incubated at 37 °C, 5% CO2, 95% humidified atmosphere. By repeated addition of topotecan or vinblastine at the maximum tolerated dose to the cell medium of MCF-7 and KB-V1 cells, the cells were rendered multidrug-resistant, indicated as MCF-7Topo and KB-V1Vbl, respectively. They were serially passaged following trypsinization by using 0.05% trypsin/0.02% EDTA (Biochrom). Mycoplasma contamination was routinely monitored, and only mycoplasma–free cultures were used.
4.7.2. MTT Assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (ABCR) was used to determine the cytotoxicity of test compounds as previously described [
53]. Briefly, HDFa human dermal fibroblasts and Ea.Hy926 endothelial hybrid cells (both 1 × 10
5 cells/mL, 100 µL/well), 518A2 melanoma, KB-V1
Vbl cervix carcinoma, MCF-7
Topo breast carcinoma, HT-29, DLD-1 and HCT-116 colon carcinoma (all 5 × 10
4 cells/mL, 100 µL/well) were grown in 96-well culture plates for 24 h. Then, various concentrations of the test compounds were added and the cells were incubated for 24-72 h at 37 °C. After adding 12.5 µL of a 0.5% MTT solution in PBS (final concentration 0.05%) to cell medium, microplates were incubated for 2 h and subsequently swiftly turned to discard the medium. The precipitate of formazan crystals was then dissolved in a 10% solution of SDS in DMSO containing 0.6% acetic acid. To ensure complete dissolution of formazan, microplates were incubated for at least 4 h at 37 °C. Finally, the absorbance at λ = 570 nm (formazan) and 630 nm (background) was measured using a microplate reader (Tecan F200). All experiments were carried out in quadruplicate and the percentage of viable cells was calculated as the mean ± SD with controls set to 100%. Selected graphs of MTT assays can be found in the
Supporting Information (
Figure S8).
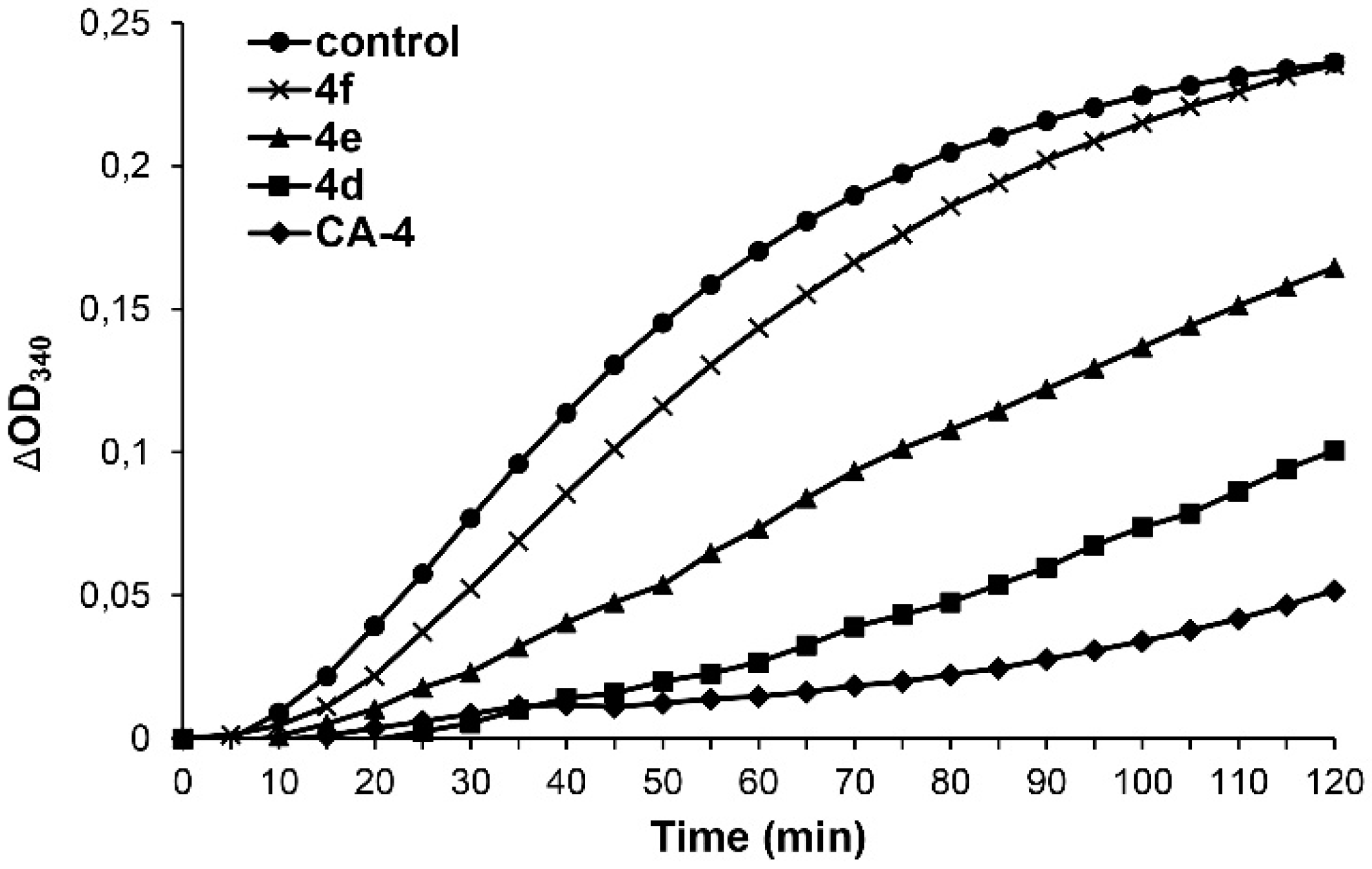
4.7.3. Tubulin Polymerization Assay
An amount of 50 µL of Brinkley’s buffer 80 (BRB80) supplemented with 20% glycerol and 3 mM GTP was given in a black 96-well half-area plate with clear bottom. Then, test compounds 4d–f (final concentration: 5 µM, or 10 µM) or vehicle (DMSO) were added. After adding 50 µL tubulin in BRB80 (10 mg/mL) was pipetted in the wells and immediately placed in a pre-heated microplate reader (Tecan). The polymerization was determined turbidimetrically at 37 °C by measuring the absorption at 340 nm for 120 min in intervals of 5 min. Values were normally distributed for each group (Shapiro–Wilk test, p > 0.05) and there was homogeneity of variance (Levene’s test, p > 0.05). The one-tailed Dunnett post-hoc test revealed significant inhibition of tubulin polymerization (p ≤ 0.001) for CA-4 and 4d compared to controls.
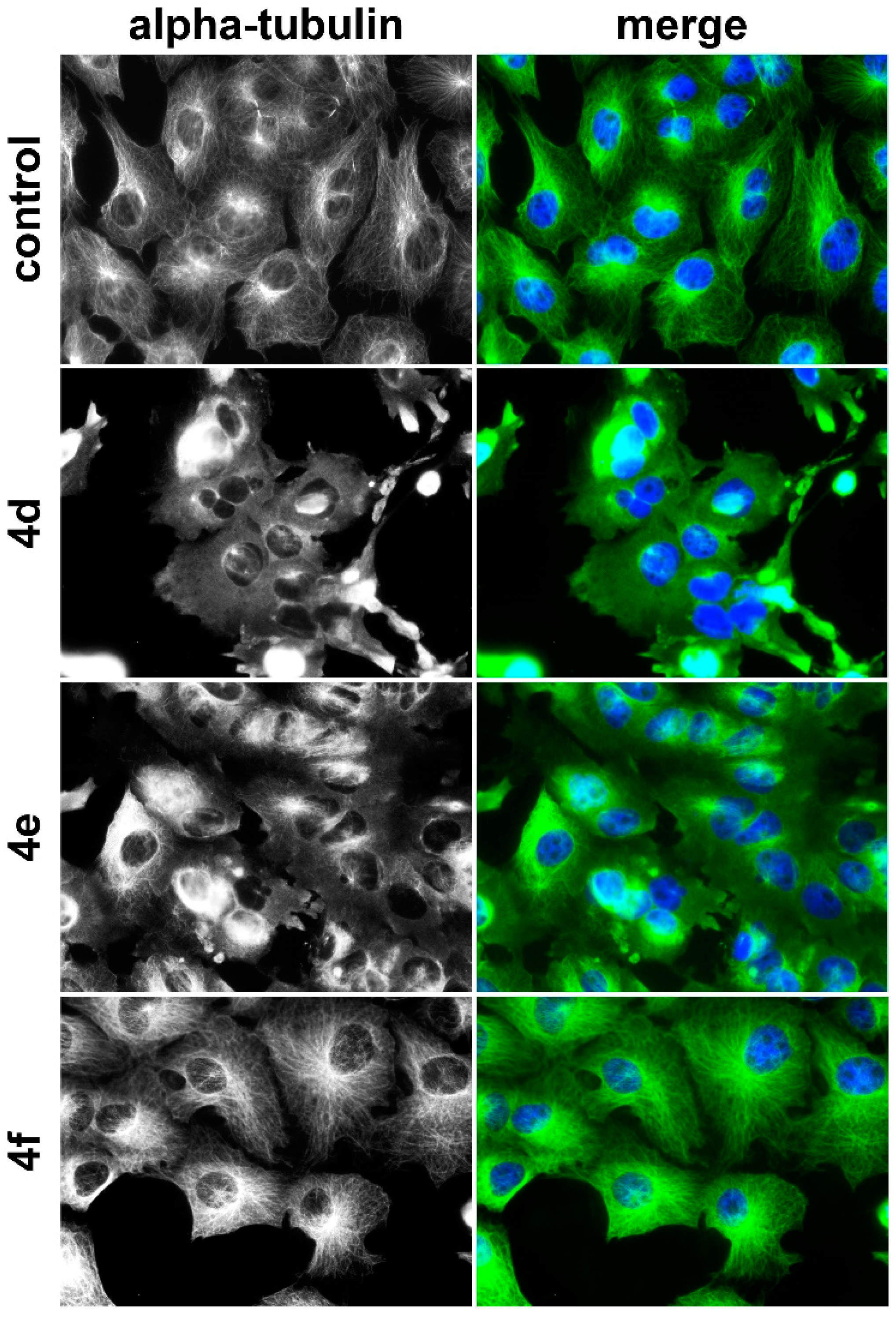
4.7.4. Immunofluorescence Staining of Microtubule Cytoskeleton
518A2 melanoma cells (5 × 104 cells/mL, 500 µL/well) or Ea.Hy926 endothelial hybrid cells (7.5 × 104 cells/mL, 500 µL/well) were seeded in 24-well plates on small glass coverslips and grown for 24 h. Then, the cells were exposed to the test compounds for 24 h. After washing the cells once with PBS, they were fixed in 3.7% formaldehyde in PBS (20 min, rt), and permeabilized and blocked in 1% BSA and 0.1% triton X-100 in PBS (30 min, rt). Then, the cells were incubated with monoclonal mouse anti-alpha-tubulin antibody (1 h, 37 °C). After washing the cells for three times with PBS, the cells were exposed to the secondary anti-mouse 488 antibody conjugate (1 h, rt, in the dark). Then, the cells were washed for three times with PBS and once with water. The glass coverslips were mounted in 4-88 based mounting medium supplemented with 1 µg/mL DAPI for counterstaining the nuclei and 2.5% DABCO. Alterations of the microtubule were documented by a Zeiss Imager A1 AX10 fluorescence microscope (400× magnification).
4.7.5. HDAC Inhibition
The HDAC inhibitory potential of the novel compounds was determined by utilizing the deacetylase activity of recombinant human HDAC1 (HDAC1 Inhibitor Screening Assay Kit; Cayman Chemicals) or HDAC6 (HDAC6 Inhibitor Screening Kit (Fluorometric); Biovision) towards the corresponding synthetic acetylated-peptide substrates resulting in the release of a fluorescent product. The assays were performed according to manufacturer’s description of the in commercially available assay kits. The fluorescence intensity (HDAC1: λex = 352 nm, λem = 452 nm; HDAC6: λex = 380 nm, λem = 510 nm) as a measure of enzyme activity was measured at 37 °C with a microplate reader (Tecan). The IC50 values were derived from dose-response curves and are expressed as the means ± SD of two independent experiments. A two-tailed t-test was performed, revealing significant differences (p < 0.0001) in HDAC6-inhibition for 4e–f compared with 4d as well as for 4d and 4f compared with 4e and in HDAC1-inhibition for 4d–e compared with 4f. The difference in HDAC1 IC50 values of 4d compared with 4e were not significant.
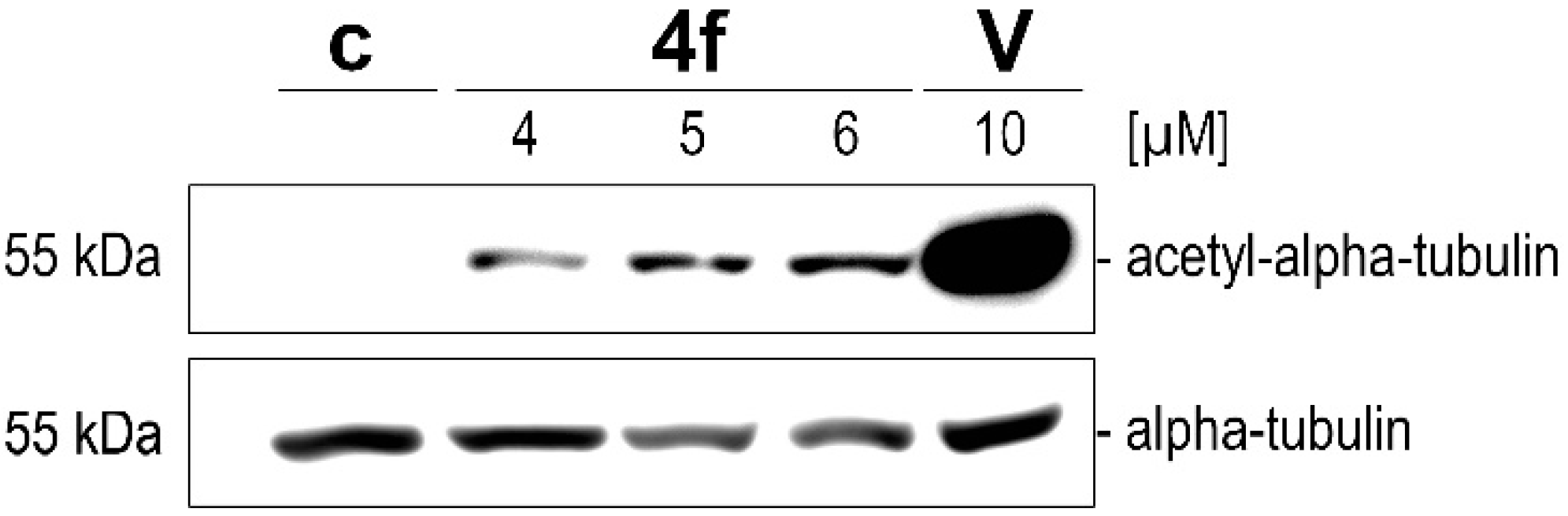
4.7.6. Western Blot Analyses
For the microtubule acetylation blots, 518A2 melanoma cells (5 × 104 cells/mL, 3 mL/well) were grown in 6-well plates for 24 h and then incubated with vehicle (DMSO), SAHA (10 µM), or 4f (4, 5, and 6 µM) for 24 h. After harvesting the cells by trypsination, they were pelleted by centrifugation (300× g, 5 min) followed by cell lysis in 100 µL lysis buffer (20 mM DTT, 200 µM sodium vanadate, 50 mM Tris/HCl, 1% triton X-100, 150 mM NaCl, pH 7.4) for 10 min on ice. The cell lysates were mixed with 100 µL of 2× Laemmli buffer (125 mM Tris-HCl, 4% SDS, 20% glycerol, 10% β-mercapto-ethanol, pH 6.8) and boiled (95 °C, 10 min). Equal amounts of total protein were subjected to 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (PVDF, Carl Roth). For subsequent analysis of acetylated protein and of alpha-tubulin which was used as a loading control, membranes were blocked and incubated with primary antibody solutions in 5% bovine serum albumin in 1× TBS or 5% milk powder in PBS, respectively. The protein bands were visualized by chemoluminescence (secondary antibody-HRP conjugates, ECL detection system; Cell Signaling) using a LAS-3000 imager (Fujifilm).
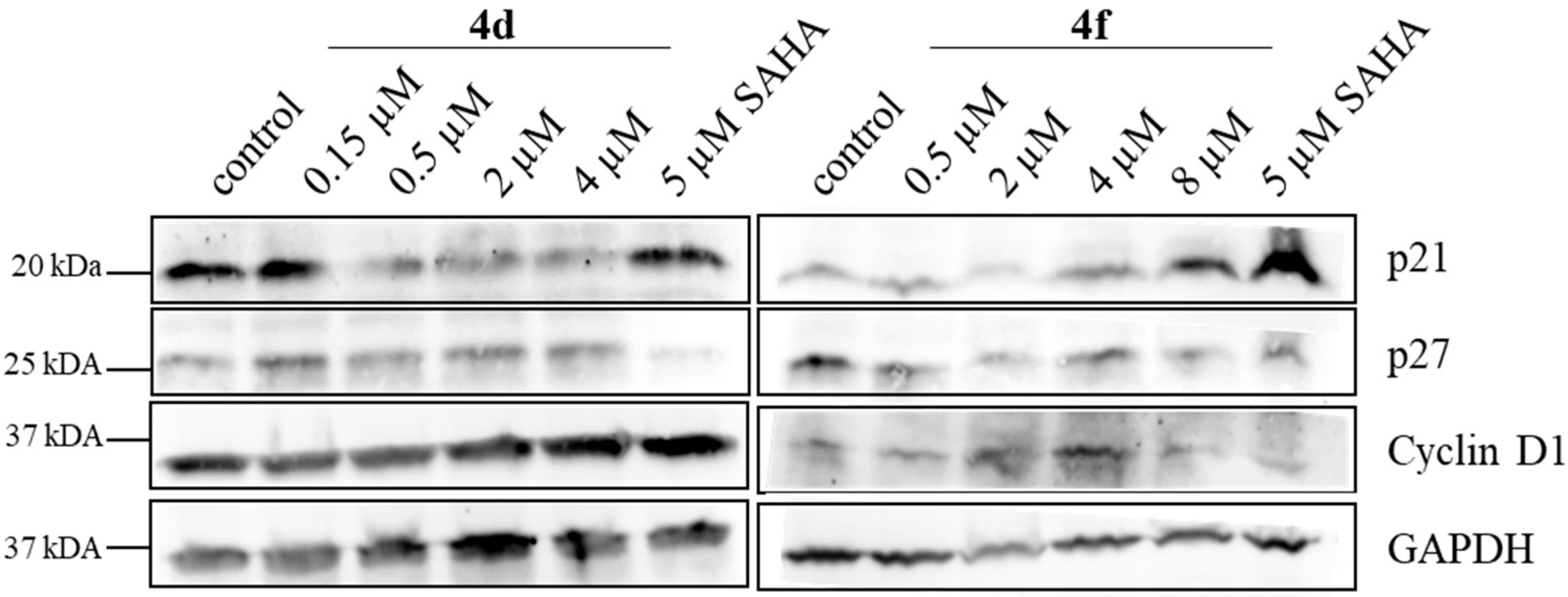
For the cell cycle protein blots, 518A2 melanoma cells (2 × 106 cells/mL, 10 mL/well) were grown in 10 cm dishes for 24 h and then incubated with
4d (0.15–4 µM) or SAHA (5 µM) for 24 h. After harvesting and lysing of the cells, western blotting was performed as described before [
54]. For subsequent analysis, the protein-loaded membranes were incubated with antibodies directed against p21 (Abcam ab109199), p27 (Abcam ab109199), cyclin D1 (Cell Signaling 29225) and GADPH (Santa Cruz sc25778) in 1:1000 dilutions. After incubation with horseradish peroxidase-coupled anti-IgG antibodies (1:10,000; Amersham), the blot was developed using the Celvin-S developer (Biostep) and the software SnapAndGo 1.8.1.
4.7.7. Immunofluorescence Staining of F-Actin
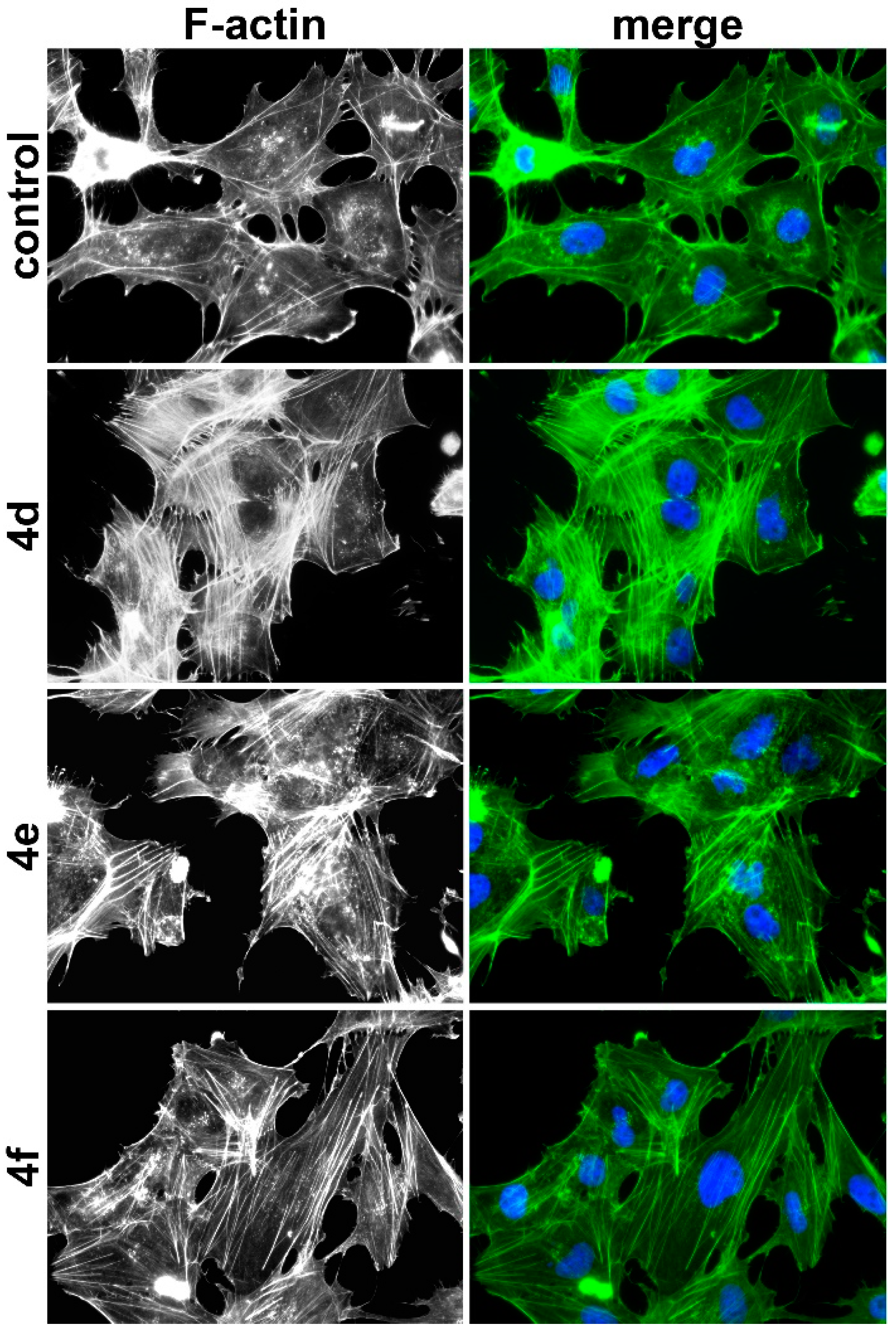
518A2 melanoma cells (5 × 104 cells/mL, 500 µL/well) were seeded in 24-well plates on small glass coverslips and grown for 24 h at 37 °C. Then, the cells were treated with 4d (0.5 µM), 4e (1.5 µM), 4f (4 µM), and vehicle (DMSO) for 24 h. After washing the cells once with PBS at 37 °C, they were fixed with 3.7% formaldehyde solution in PBS (pH 7.0) for 10 min at room temperature. Then, the cells were washed with PBS and permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. After washing with PBS, the cells were incubated with 200 µL of a 0.1 µM Acti-stainTM 488 phalloidin solution in PBS for 30 min at room temperature in the dark. Then, the cells were washed three times with PBS and once with water, and the glass coverslips mounted in 4-88-based mounting medium containing 1 µg/mL DAPI for counterstaining the nuclei and 2.5% DABCO. The effects on the actin cytoskeleton were documented by a Zeiss Imager A1 AX10 fluorescence microscope (400× magnification).
4.7.8. Cell Cycle Analysis
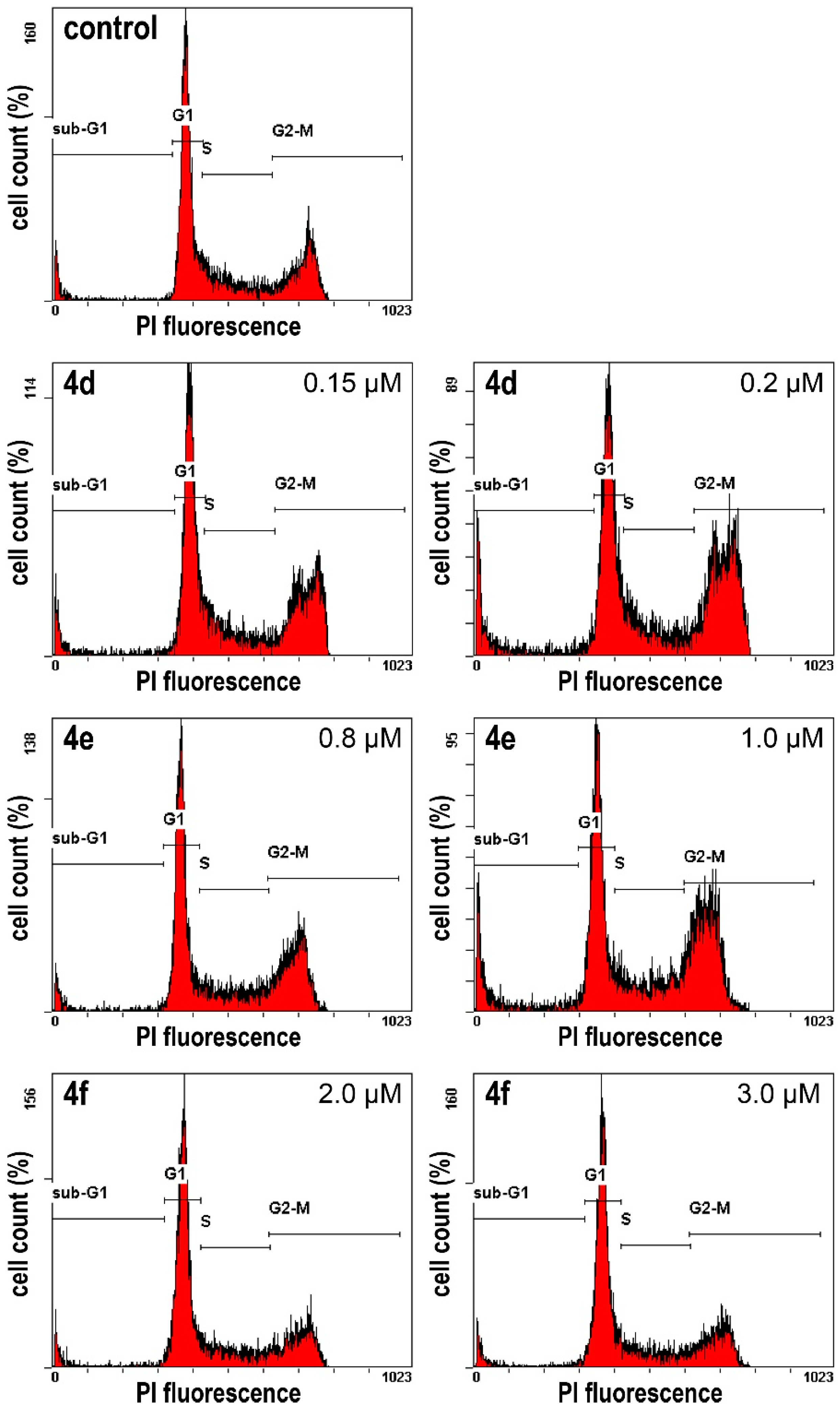
The 518A2 melanoma cells (3 mL/well; 5 × 104 cells/mL) were grown on 6-well tissue culture plates for 24 h. After treatment with 4d (150, and 200 nM), 4e (0.8, and 1 µM), 4f (2, and 3 µM), or DMSO (control) for another 24 h, cells were fixed (70% EtOH, 24 h, 4 °C), washed with PBS and incubated with propidium iodide (PI; Carl Roth) staining solution (50 µg/mL PI, 0.1% sodium citrate, 50 µg/mL RNase A in PBS) for 30 min at 37 °C. The fluorescence intensity of 10,000 single cells was measured at λem = 570 nm (λex = 488 nm laser source) with a Beckmann Coulter Cytomics FC 500 flow cytometer. The percentages of cells in the different phases of the cell cycle (G1, S and G2/M phase) were determined using the CXP Analysis software (Beckmann Coulter). The percentage of apoptotic cells was derived from sub-G1 peaks. Data was normally distributed for each group (Shapiro–Wilk test, p > 0.05). Since homogeneity of variance (Levene’s test, p < 0.05) was violated, a correction was calculated for Anova (Welch test). The two-tailed Games–Howell’s post-hoc test revealed the significant alteration (p ≤ 0.05) of the population of cells in G1 in 518A2 melanoma cells treated with 4d–f compared with vehicle-treated controls.
4.7.9. In Vivo Toxicity
In vivo toxicity of 4d was studied in nude mice (Charles River Laboratories, Sulzfeld, Germany). These experiments were carried out following the institutional guidelines. 4d was formulated in 10% Tween80/10% ethanol/80% saline for administration. One mouse (32 g) was treated once with 1 × 100 mg/kg body weight (i.p.), another mouse (30 g) was treated once with 1 × 200 mg/kg body weight (orally) of 4d and then both mice were observed for two weeks. The body weight of the mice was assessed daily under therapy.
4.8. In Silico Evaluation
4.8.1. Proteins and Compound Structures
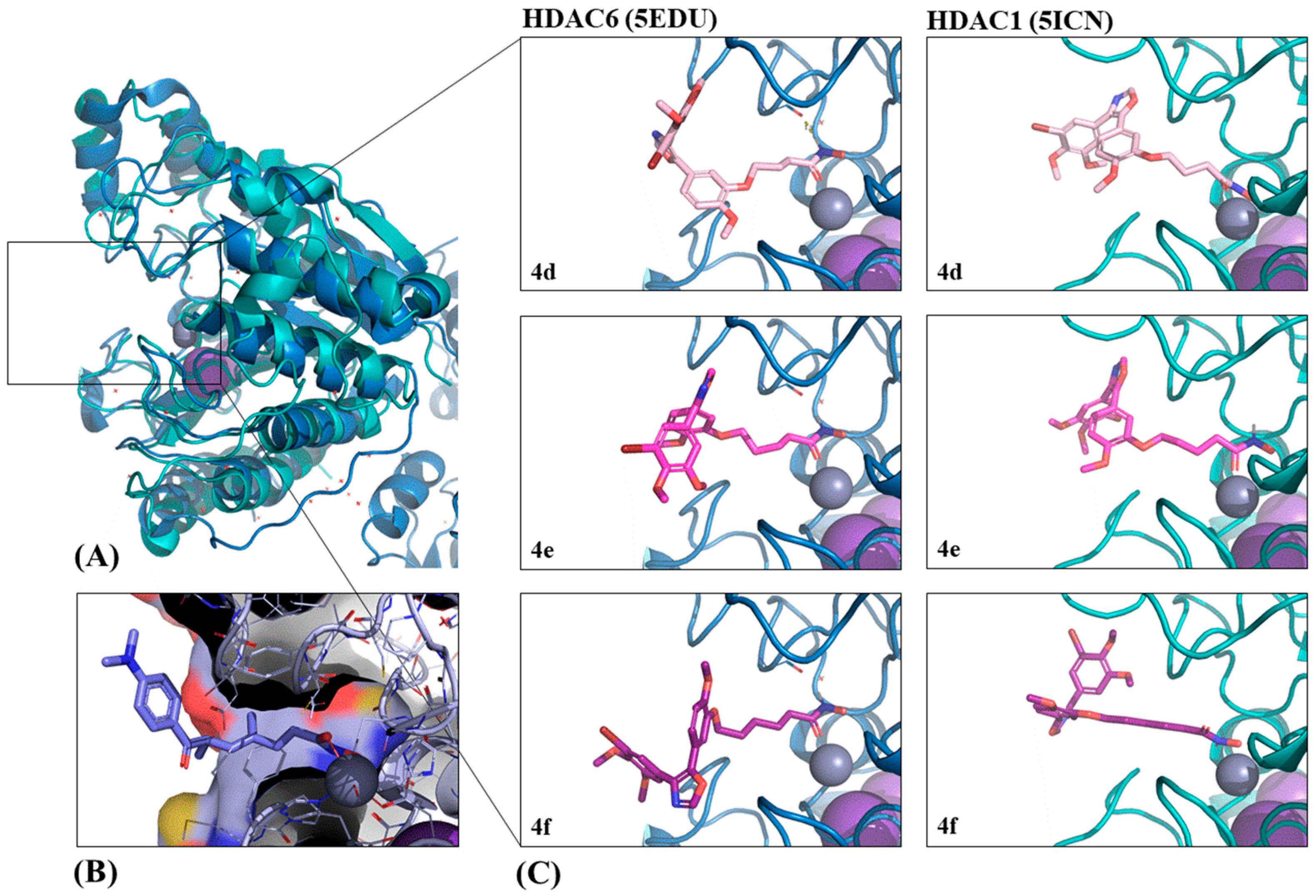
The protein structures for tubulin, HDAC1, and HDAC6 used in the docking studies were obtained from the Protein Data Bank (
Table 7, PDB,
www.rcsb.org, accessed 6 March 2018) [
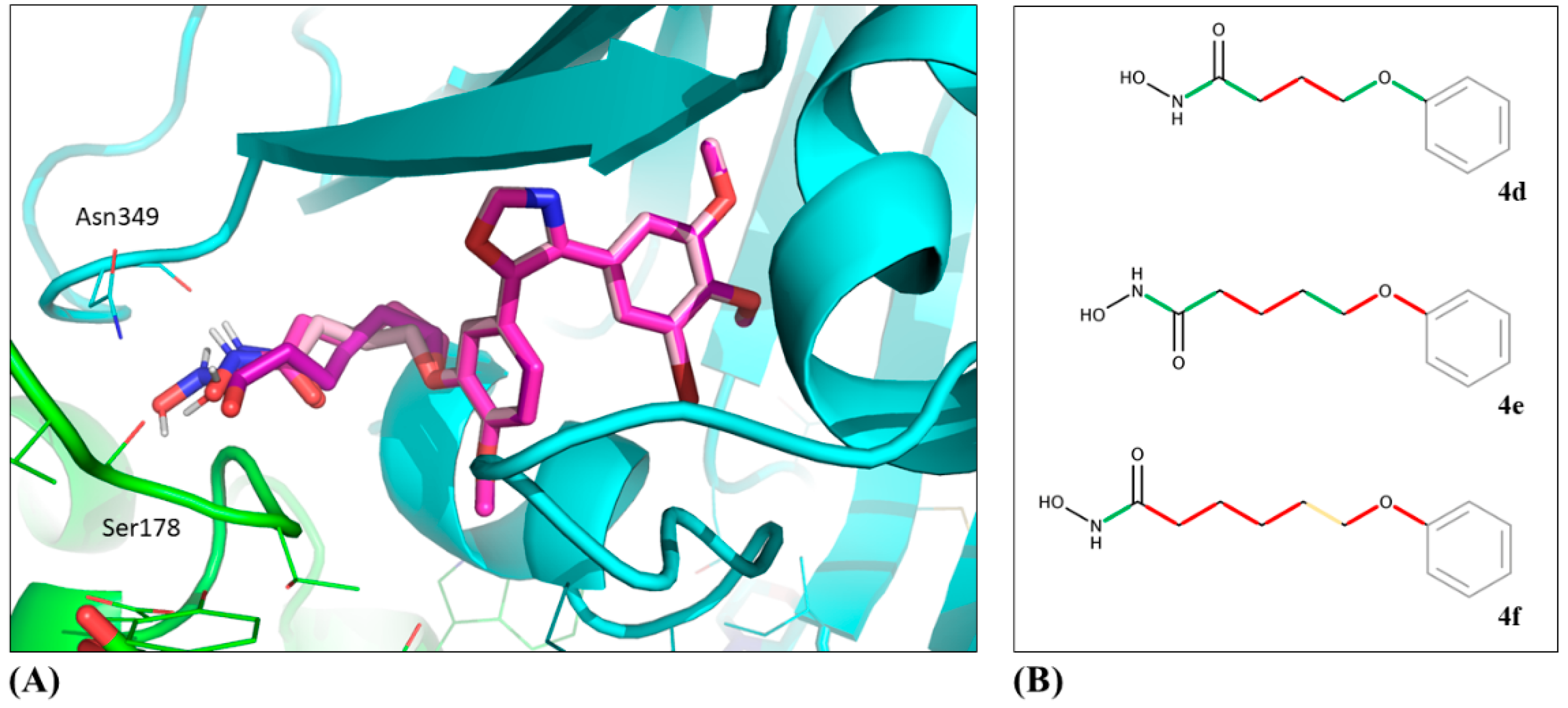
55]. One representative per protein was selected for the docking studies based on the following criteria: The protein should be co-crystallized with a ligand most similar to the ligands from which the studied hybrid compounds
4d–
f were derived (combretastatin A-4 for tubulin and hydroxamic acids for HDACs) [
56]. Among these, the structures with the best resolutions were selected.
4.8.2. Initial Docking with LeadIT/FlexX
To generate initial predictions of binding positions, called docking poses, FlexX (included in LeadIT 2.2.0, which was kindly provided by BioSolveIT,
www.biosolveit.de/LeadIT), was used [
57]. The PDB structures were prepared as follows: For tubulin, chains A and B of 5LYJ, for HDAC1, chain B of 5ICN, and for HDAC6, chain A of 5EDU were chosen [
47,
50,
51]. The binding pockets were defined with the co-crystallized ligands as a reference ligand, including all amino acids within a radius of 10 Å for the HDAC structures and 6.5 Å for tubulin. The metal coordination of the zinc ion in both HDAC structures was set to ’spherical’ and zinc was defined as an essential pharmacophore for guided docking. All other settings for the receptor definition were used as default. The docking library contained compounds
4d–
f as well as the crystal structure’s original ligand. For HDAC1 and HDAC6 vorinostat, a known HDAC inhibitor, was added to the library. The docking strategy, scoring and chemical parameters were kept as default. Only the maximum number of solutions per iteration and the maximum number of solutions per fragmentation were increased to 1000 steps each.
4.8.3. Pose Optimization with SeeSAR
Final pose optimization and affinity estimation was performed with SeeSAR 6.1, also kindly provided by BioSolveIT (
www.biosolveit.de/SeeSAR). The best poses from the LeadIT docking were imported and were used for binding site definition. For each compound, 10 new poses were generated, and all were evaluated with the built in HYDE scoring function [
58]. The poses with the best estimated affinities were chosen for further analysis.
4.8.4. Additional Software Used
PyMOL was used to analyze, compare, and visualize the binding pose predictions, as well as to create the 3D images [
59].

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}