1. Introduction
Calcium phosphate bone cements (CPCs) have been of interest in the field of bone regeneration for over 30 years. CPCs are hydraulic cements composed of calcium orthophosphate which forms a mouldable paste following mixing with a liquid phase generally consisting of pure water or aqueous sodium phosphate, physiological saline or phosphate buffered saline [
1]. CPCs are also tagged as self-setting materials due to their ability to harden in vivo via a chemical setting reaction under physiological conditions [
2,
3]. One of the main positive aspects of CPCs consists of their injectability which allows for the cement to be implanted by means of mini-invasive surgical procedures. Thus, they have significant potential with respect to the fields of orthopaedics, traumatology (fracture fixation) and dentistry. However, commercial CPCs do not yet feature all the desired properties; in particular, they are not completely injectable at low force levels and with high solid contents without the occurrence of separation, and demonstrate washout resistance to body fluids soon after mixing. Supplementing CPCs with polymeric additives in different forms appears to provide a promising strategy for the avoidance of the afore-mentioned limitations. A number of polymers have been explored with respect to improving the properties of CPCs, such as natural hydrogels based on collagen [
4,
5], gelatine [
6], carboxymethylcellulose [
7], hyaluronic acid [
7], a hyaluronan-bisphosphonate composite [
7], chitosan [
8], synthetic water-soluble poly (ethylene glycol) [
9] and biodegradable poly(propylene fumarate) crosslinked with hydroxyethyl methacrylate [
10]. Moreover, a further, novel CPC design involving a thermo-responsive copolymer known as Poloxamer has recently been demonstrated [
11]. Poloxamer, a triblock copolymer of poly (ethylene glycol-
b-propylene glycol-
b-ethylene glycol) [
12], thermogels at body temperature, thus increasing the viscosity of the CPC immediately following injection into the site of the defect and enhancing washout resistance [
11]. However, Poloxamer is not biodegradable, it is soluble only in physiological fluids and exhibits toxicity at higher concentrations [
12].
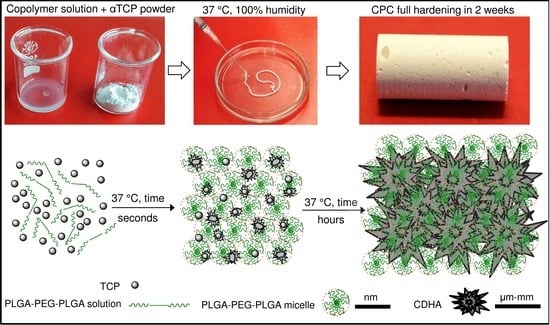
This study investigates a biodegradable, non-toxic, thermosensitive copolymer based on FDA-approved poly(lactic acid), poly(glycolic acid) and poly(ethylene glycol) (PLGA–PEG–PLGA), commercially known as the ReGel
® or OncoGel
® drug delivery systems incorporating insulin and the drug Paclitaxel, respectively. In a similar way to Poloxamer, the PLGA–PEG–PLGA copolymer forms a free-flowing sol at room temperature and a physical three-dimensional network hydrogel at body temperature resulting in very good injectability due to its thixotropic rheological behaviour [
13]. Moreover, PLGA–PEG–PLGA is fully biodegradable in the Krebs cycle to carbon dioxide and water [
14]. However, the use of PLGA–PEG–PLGA as a drug delivery system is limited, since the copolymer exhibits a low degree of functionality; hence, a great deal of attention has been devoted to PLGA–PEG–PLGA functionalization [
15,
16]. Recently, we modified the PLGA–PEG–PLGA copolymer via the addition of inorganic bioactive hydroxyapatite (HA) in the form of micro-, nano- and core-shell particles [
17] and the results revealed that these composites inherited the sol-gel and gel-sol phase transitions of the polymer solutions. These characteristics subsequently led to the assumption that the addition of the PLGA–PEG–PLGA copolymer may provide for the formation of a new class of CPCs with properties closer to the ideal.
The aim of this study, therefore, was to assess the effect of a thermo-sensitive PLGA–PEG–PLGA copolymer on the setting reaction, injectability and cohesive properties of a CPC that sets due to the hydrolytic transformation of alpha-tricalcium phosphate (α-TCP) into calcium deficient hydroxyapatite (CDHA) according to the following chemical reaction (1).
Emphasis was placed on correlating the time-dependent rheological behaviour of the cement with the kinetics of the setting reaction, injectability and washout resistance. Finally, the degradation behaviour and cytocompatibility of the cement were assessed in views of its potential application for bone regeneration purposes.
3. Discussion
In general, the characterization of the visco-elastic properties of CPCs is a complex task due to the influence of the setting reaction [
28]. In addition, no method is available for the determination of the setting of the cement in correlation to the evolution of the hydrolysis reaction (Equation (1)). Commonly, the setting time is determined by means of a mechanical method based on the indentation of one or two needles on the surface of the cement paste. Although these methods allow for a comparison of differing cement formulations, i.e., once they achieve certain strength, they do not directly correlate with the advancement of the hydrolysis reaction. Other methods that allow for the determination of the crystalline composition, such XRD, are not appropriate with respect to follow the initial setting of the cement since at very short time points, the changes in the crystalline composition are below the XRD detection limit. Rheological measurement provides the relevant information on the visco-elasticity of the pastes, as well as the setting process that reflects the evaluation of the internal (polymer or ion concentration, particle size, distribution, shape) and external (temperature) conditions during the self-hardening process [
29]. It should be borne in mind that the setting reaction, shown in Equation (1), is based on the dissolution of α-TCP and the precipitation of CDHA [
30]. Immediately following mixing, the dissolution of α-TCP regulates the process up to the commencement of the precipitation of the CDHA, from which point the dissolution of the α-TCP and the growth of new CDHA crystals progress in parallel. With concern to this mechanism, it is difficult to define when exactly the cement sets. Moreover, the dissolution rate of α-TCP depends on the environment [
21]. Consequently, this study considered the use of ultrapure water only, i.e., without the presence of other inorganic additives, in order to gain a better understanding of the setting mechanism.
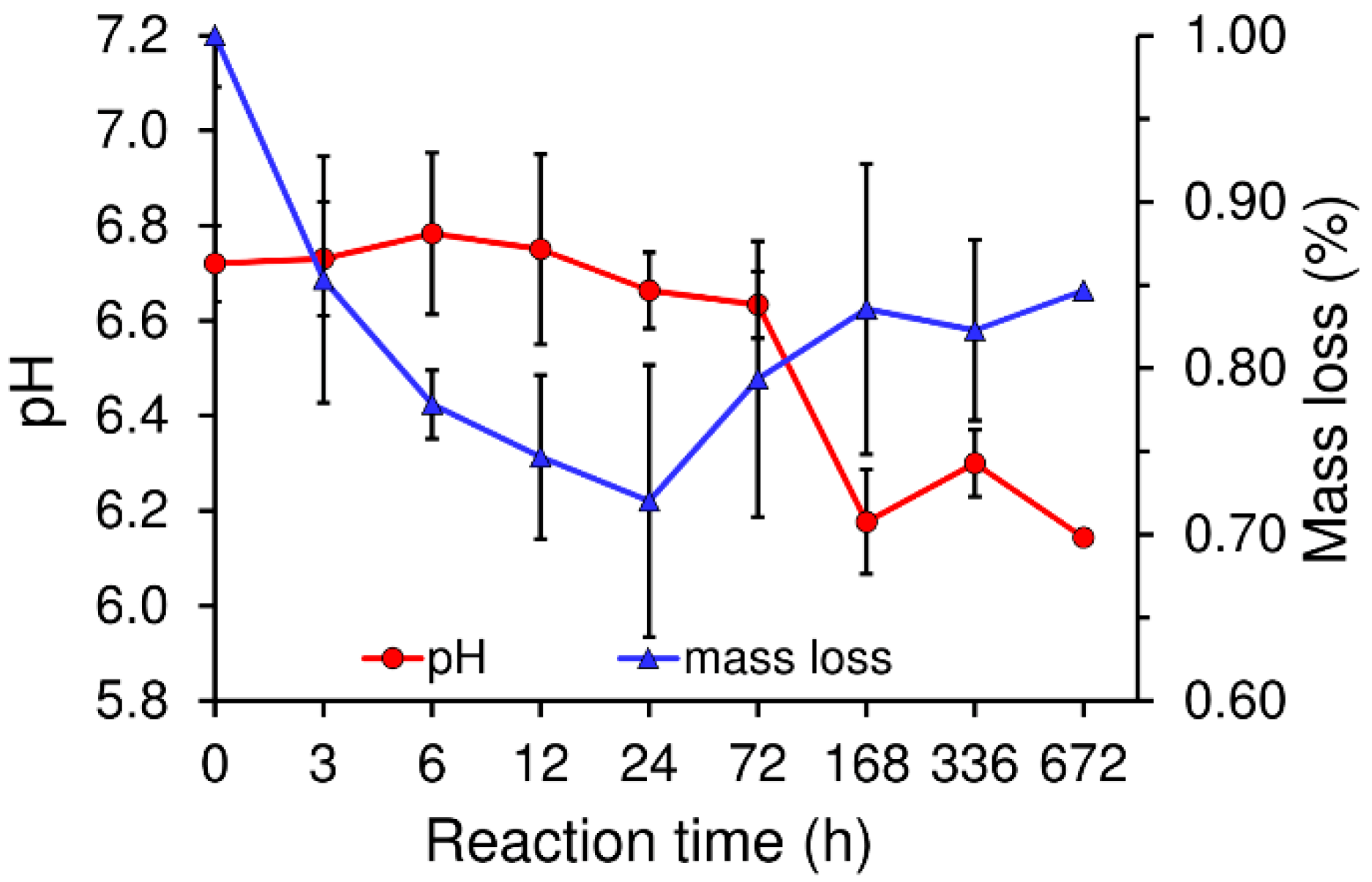
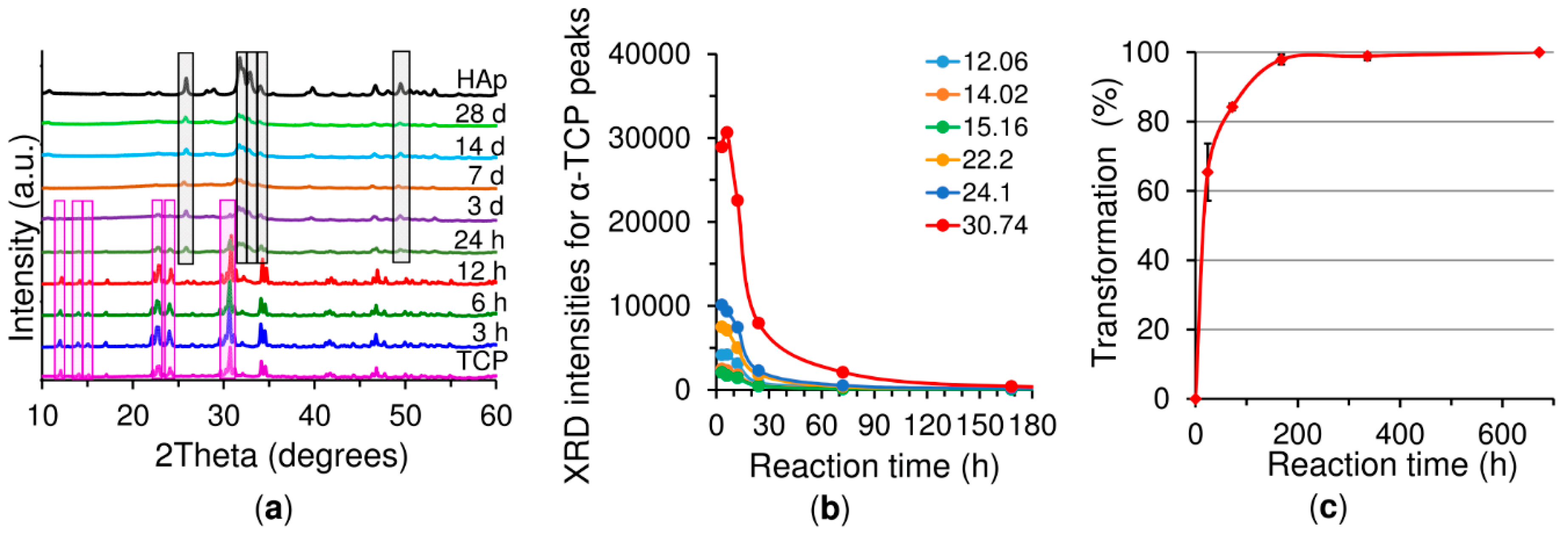
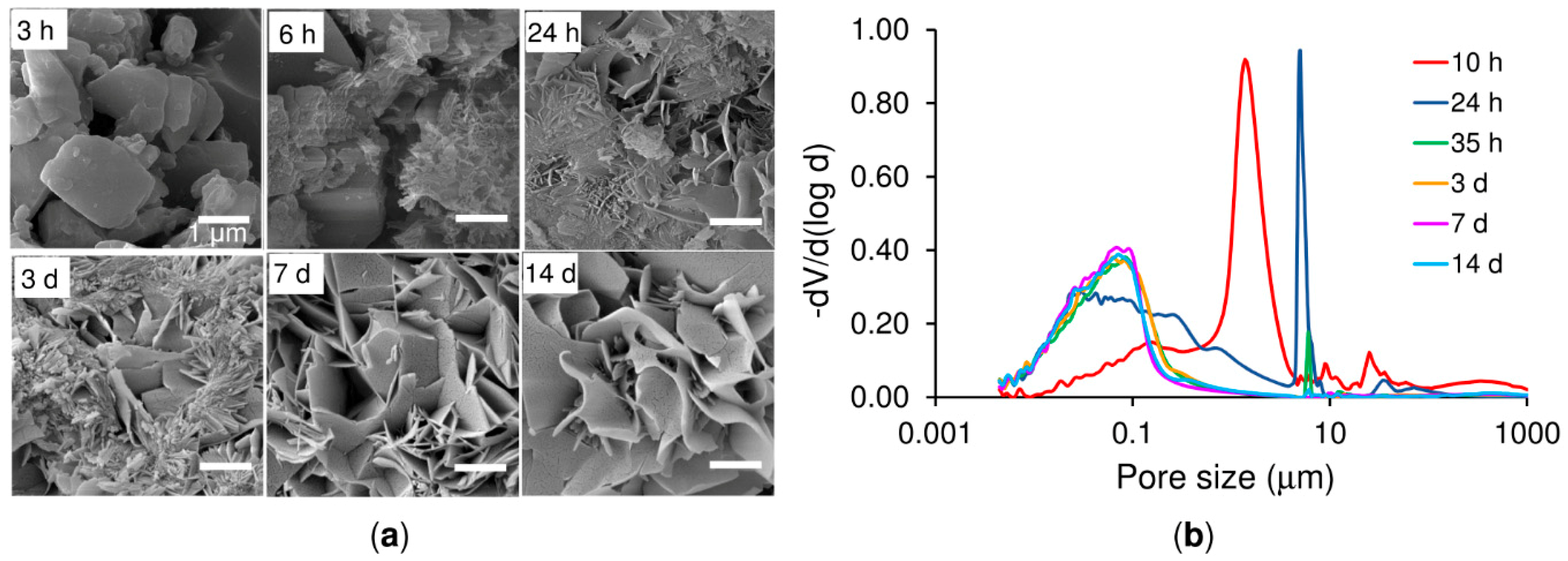
In general, setting is understood as the loss of cement paste workability, while hardening is defined as the increase in the strength of a solid cement block until a total chemical reaction is achieved. The question then is how to correlate workability with the chemistry. The microstructural analysis determined that the first visible CDHA crystals appeared after 6 h of reaction on the surface of the α-TCP particles. Nonetheless, CDHA diffraction peaks were not observed until after 12 h of reaction. Nevertheless, XRD provided information on the dissolution of the α-TCP particles in that the increase in the intensity of the diffraction peaks of the α-TCP could be explained by the dissolution of the most reactive domains of the α-TCP particles. After 6 h of reaction this trend was reversed with the occurrence of a decrease in the intensity of the diffraction peaks of the α-TCP that correlated with the time at which the first CDHA crystals were observed within the microstructure of the cement, thus possibly representing nucleation and the commencement of the growth of the CDHA crystals. The nucleation of the CDHA crystals after 6 h of reaction was further supported by the variation in the pH in the water in which the cement set. The pH increased up to a maximum value after 6 h of reaction due to the release of ions from the cement particles (
Figure 4). Subsequently, and in correlation with the nucleation of the CDHA crystals, the pH decreased, since the nucleation and growth of the CDHA required the consumption of calcium, phosphate and hydroxyl ions. The pore size distribution (
Figure 6b) also reflected the changes in the microstructure during the α-TCP reaction. Firstly, the pore size between the α-TCP particles increased due to the dissolution thereof; however, the volume of the pores decreased (thinner peak) due to the growth of the CDHA crystals. Finally, the spaces between the particles were completely filled by the CDHA crystals, resulting exclusively in pores of less than 1 µm in size.
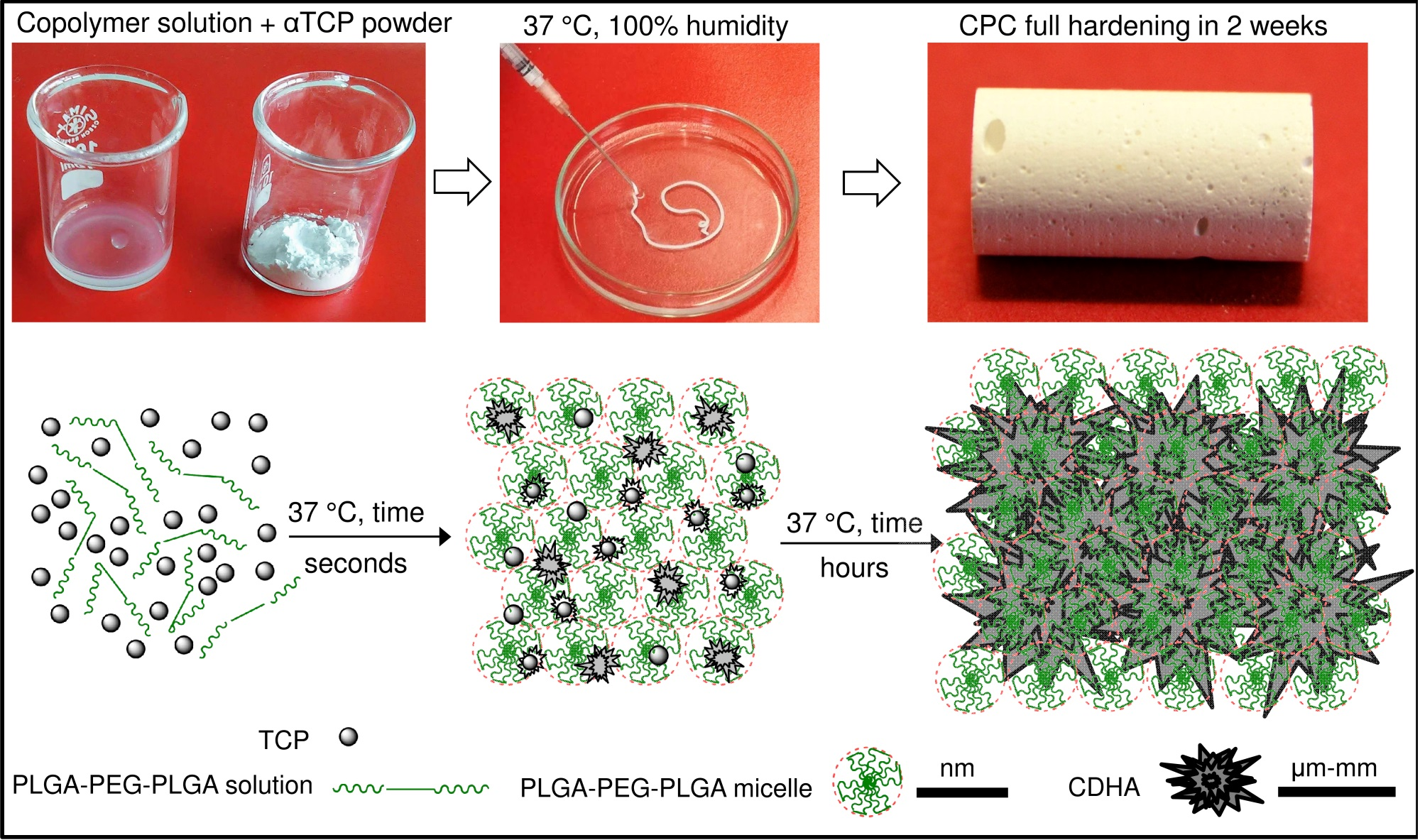
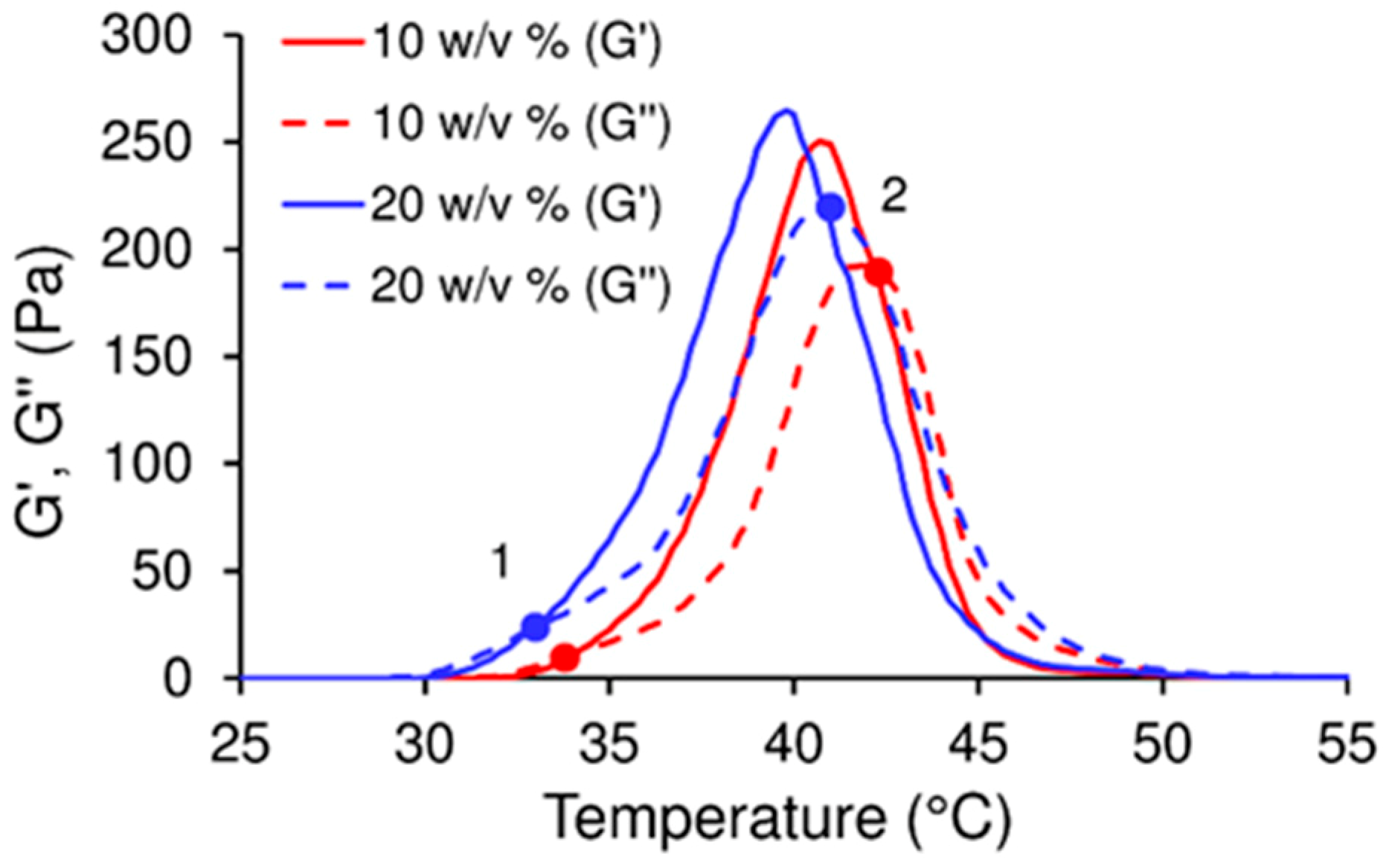
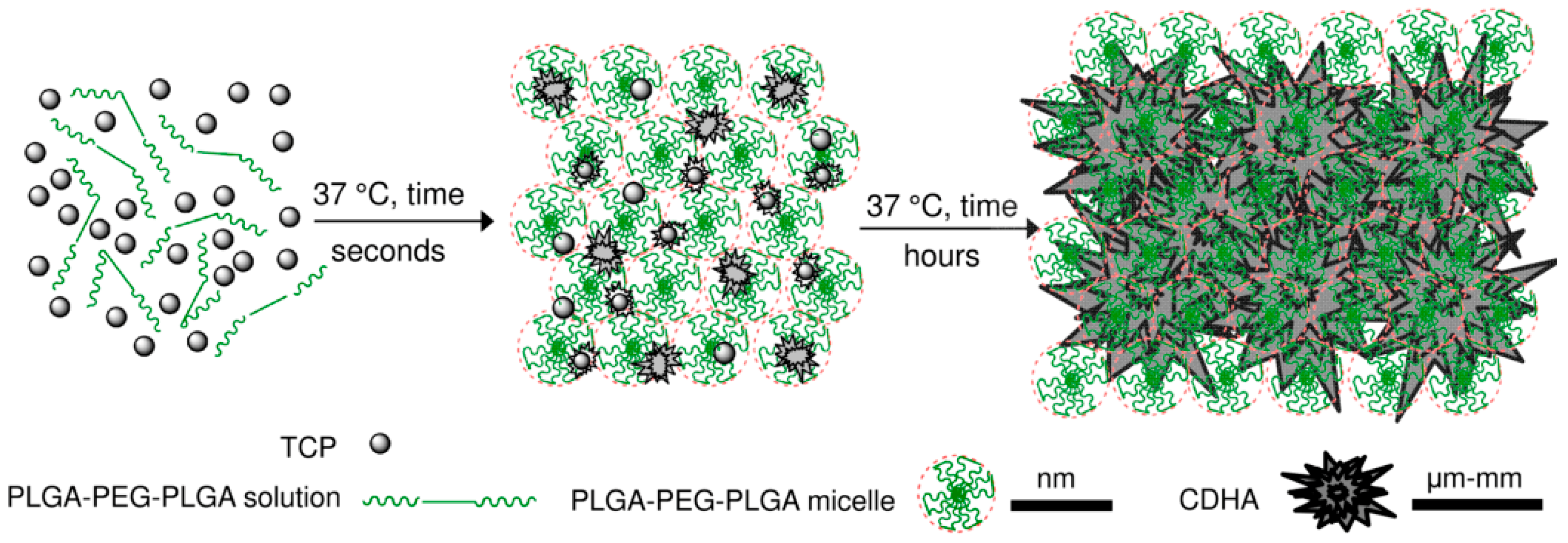
Previous analysis demonstrated that the nucleation of CDHA commenced after 6 h of the reaction between the copolymer water solution and the α-TCP; however, the means by which the workability of the cement changed during this time was addressed solely by means of time-sweep curves of the cement, which demonstrated a sharp increase in the storage modulus thus indicating a change in the microstructure and nucleation development of the CDHA. Prior to the sharp increase in the storage modulus (within the lag phase), the cement pastes exhibited, in general, a constant storage modulus, the magnitude of which depended on parameters, including the L/P ratio, copolymer content and temperature. It is a general concept that temperature significantly affects the storage modulus of thermo-sensitive copolymers; such copolymers convert from the liquid to the gel state with an increase in temperature via micelle-packing and gel formation [
18]. Moreover, the entire self-setting process is predominantly accelerated by temperature [
20]. However, as the scheme in
Figure 10 shows, while the gelation of copolymers is rapid (a few seconds), the setting of α-TCP takes a number of hours to several days. Therefore, as shown in
Figure 2, the stiffness of cement pastes prior to CDHA nucleation is controlled by the degree of gelation of the copolymer which, in turn, directly depends on the concentration of the copolymer and the temperature.
The effect of temperature on the setting reaction was observed as a shorter lag phase or, in other words, the more rapid nucleation of the CDHA due to temperature promoting the dissolution of α-TCP. The addition of the copolymer significantly reduces the storage modulus of the cement in the lag phase, since the copolymer acts as a surfactant that decreases the interparticle forces within the cement. In contrast, when mixed with water, the α-TCP particles in the cement are subjected to attractive interaction forces which act to form a stiff material. Such pastes with high solid contents are very difficult to inject due to phase-separation [
31,
32]. It should be noted that when mixed with the copolymer, the nucleation of the CDHA was clearly observed as a sharp increase in the storage modulus, whereas when mixed with water no well-defined change in the modulus was observed. It should be possible via the conducting of a longer rheological experiment to observe the increase in the storage modulus of cements mixed with water, in a similar way to the increase in compressive strength. Therefore, in contrast to standard methods that determine setting times via indentation, time-sweep curves are able to discern between “false” setting due to the higher compaction of the cement as caused by a decrease in the L/P ratio and “real” setting caused by the attainment of local conditions for CDHA nucleation in a shorter time. In addition, since the setting time determined by needle indentation may differ from that determined by means of time-sweep curves, a new criterion other than the 15 min of final setting time proposed by Driessens et al. [
33] will be required with respect to the selection of cements to be employed in clinical applications.
In contrast to the time-sweep curves obtained at a very low frequency and strain level, steady rheological analysis and viscosity curves obtained by the gradual increase of shear strain provide information on the workability, injectability and washout resistance of the cements. The yield stress observed in the stress-strain curves reflects the yield force required to start the injection of the cement paste [
19]. The yield shear stress for the α-TCP paste with water was 3 times higher than that of the α-TCP paste with the copolymer. The difference was due to the surfactant nature of the copolymer that acts to reduce the attractive colloidal forces between the α-TCP particles. Congruently, it was very difficult to inject the α-TCP paste with water. In contrast, the lower yield stress of the α-TCP paste with the copolymer allowed for the easy injection of the paste through a 20 G needle without the occurrence of phase separation signals.
While yield stress defines the ease with which the cement can be injected, the trend in the reduction of shear stress following maximum stress and the recovery of viscosity once the shear strain is reduced provides information on phase separation during injection and paste cohesion when immersed in liquids. On the one hand, the pastes that retained a high level of shear stress when the shear strain increased did not exhibit phase separation during either the rheological or injection tests, thus allowing for minimally invasive implantation. However, on the other, those pastes that underwent a drastic reduction in shear stress following yield stress exhibited a tendency towards phase separation.
Figure 3c,e clearly show both how the α-TCP paste prepared with water separated following rheological testing and how this is in concordance with disintegration in contact with water. In contrast, the α-TCP paste prepared with the copolymer remained homogeneous following rheological testing (
Figure 3d), which is in agreement with both ease of injection, as can be seen by the formation of a filament, and cohesion, since the filament preserves its shape in contact with water (
Figure 3f). Conversely, the high shear stress following yield together with the more rapid recovery of the viscous state prevented the occurrence of the disintegration of the paste in contact with liquids, thus providing anti-washout resistance. In other words, after placing the sample in position, the attraction interactions within the paste reform once the paste is at rest. The faster the re-formation of the attractive interactions, the shorter the cohesion time of the cement (minimum time required from mixing to observing anti-washout resistance). Note that at this time scale, the increase in attractive interactions is almost independent of the progress of the setting reaction and mainly depends on the pseudoplastic nature of the copolymer. However, a fast setting reaction contributes towards increasing the cohesion of the cement over a shorter time. In fact,
Figure 3 clearly shows how the thermo-sensitive copolymer changes the behaviour of the cement paste from extremely thixotropic to increasingly pseudoplastic. This phenomenon is thought to be due to the intrinsic pseudoplastic behaviour of the copolymer promoted by gelation upon an increase in temperature. Thus, in addition to quantitatively assessing the setting behaviour of the α-TCP, rheological testing also provides a valuable tool for the prediction of the cohesion of CPC pastes.
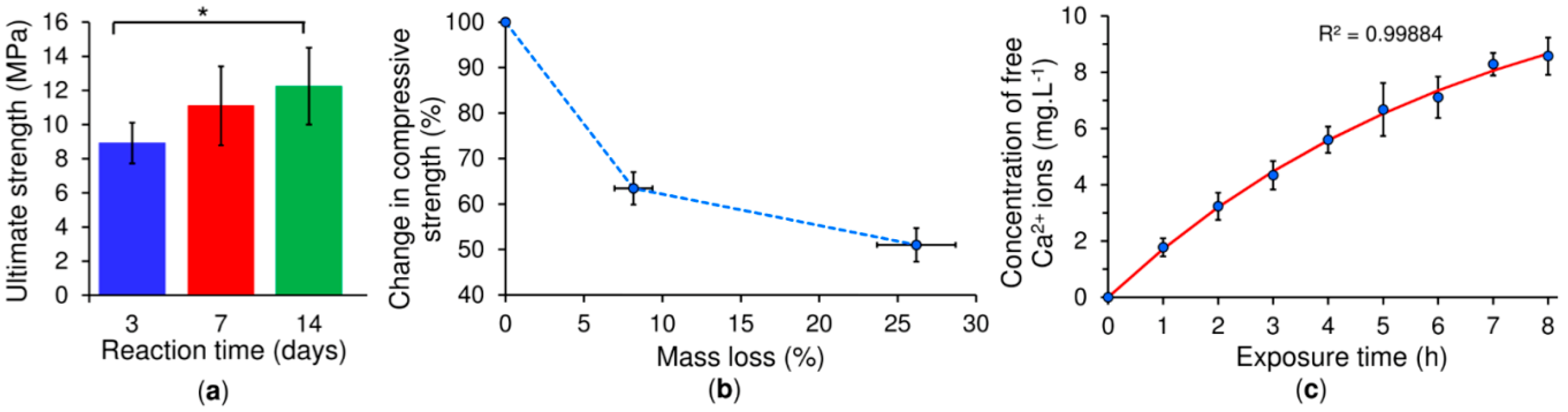
Due to the force measurement limitations inherent to rheometers, it is necessary to study the hardening of the copolymer/
α-TCP by means of a different method, commonly the compression test. Results have shown that compressive strength follows the same trend as the percentage of reaction, since the strength of the solid cement depends on the interlocking of the CDHA crystals, which increases along with CDHA growth. The accelerated degradation test was inspired by the acidic milieu attaining values of below pH 3 generated by osteoclasts in the bone resorption process and resulting in the dissolution of underlying minerals [
25]. Even though this study does not cover the complexity of the degradation that occurs in the body [
24], it does demonstrate the mass loss of the cement and the release of calcium ions over the exposure time. Moreover, the degradation of the cement generates pores on the surface with size and morphology similar to the reabsorption pits produced by osteoclasts, thus leading to a reduction in the compressive strength of the cement. A low pH is also able to catalyse the breakage of the ester linkage of the copolymer backbone and lead to higher polymer hydrolytic degradation [
34,
35]. Glycolic and lactic acid monomers produced upon PLGA degradation are further able to catalyse the dissolution of the cement [
36], which may present an advantage for this new cement formulation in that it is potentially able to overcome the long resorption times observed in vivo with respect to other cement formulations [
37]. A further approach to increasing the resorption rate of the cement consists of the incorporation of pores larger than 100 μm, which will allow for bone ingrowth and cement resorption [
38]. Interestingly, the use of the copolymer did not exhibit cytotoxicity in contact with hMSC. Indeed, the cells exhibited a typical hMSC morphology, indicating that these scaffolds exerted no notable adverse effects on cell viability in this short-term in vitro study. Since cells may detach following prolonged static culturing due to the low calcium concentration, it is planned that soon, we will employ a hydrodynamic culturing system [
39] in order to evaluate cytocompatibility over an extended culturing period. Moreover, we propose to compile an assessment of the novel copolymer/
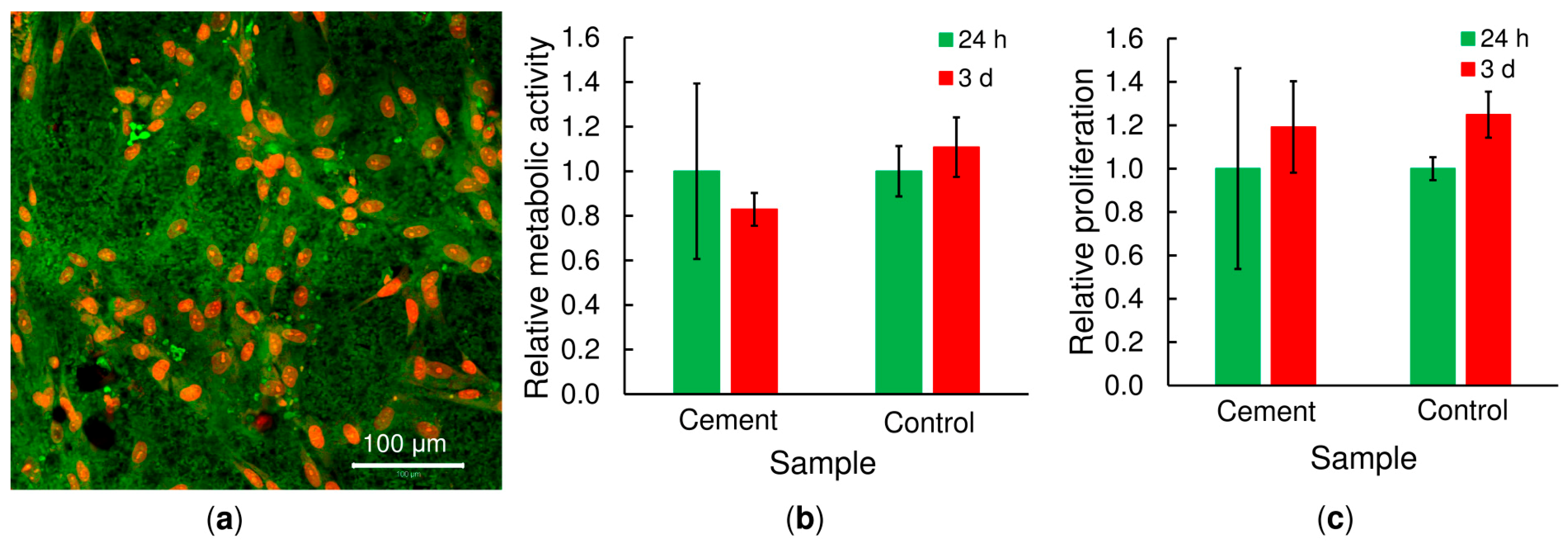
α-TCP cement in terms of normal immunological response using a large animal model (a White pig) in order to confirm its regenerative performance in vivo. The afore-mentioned properties indicate that this material presents a very promising system for use in mini-invasive surgery especially with concern to the delivery of thin-needle injection applications into bone defects and the fixation of comminuted bone fractures.
4. Materials and Methods
4.1. Chemicals
D,L-lactide (LA, ≥ 99.9%) and glycolide (GA, ≥ 99.9%) were supplied by Polysciences (Warrington, PA, USA). Poly(ethylene glycol), Stannous 2-ethylhexanoate (95%), calcium carbonate (CaCO3, ≥ 99.0%) and calcium hydrogen phosphate (CaHPO4, ≥ 98.0%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The ultrapure water (Type 1 according to ISO 3696) was prepared using the Millipore purification system (Milli-Q Academic, Millipore, Molsheim, France).
4.2. Synthesis of PLGA–PEG–PLGA Triblock Copolymers (Liquid Component of the CPC)
The PLGA–PEG–PLGA triblock copolymer with a theoretical molecular weight of 5250 g·mol
−1, an LA/GA molar ratio of 3.0 and a PLGA/PEG weight ratio of 2.5 was synthetized in a nitrogen atmosphere via the ring opening polymerization method (
Supplementary Figure S5) in a bulk according to Michlovská et al. [
18]. Briefly, a one-pot reaction of calculated D,L-lactide, glycolide, poly(ethylene glycol) and stannous 2-ethylhexanoate proceeded in the nitrogen atmosphere in a high-vacuum all-glass pipe line at 130 °C for 3 h. Any unreacted chemicals were removed by means of rinsing the product three times with 80 °C ultrapure water. The resulting freeze-dried copolymer was dissolved in ultrapure water at 4 °C thus providing 10
w/
v% and 20
w/
v% solutions.
4.3. Synthesis of α-Tricalcium Phosphate (Powder Component of the CPC)
A well-established method was used to synthetize the
α-TCP at 1400 °C via a solid-state reaction from a 2:1 molar mixture of CaCO
3 and anhydrous CaHPO
4 followed by air quenching so as to prevent the formation of the
β-TCP polymorph [
19]. The block obtained was dry milled for 15 min at 450 rpm in a planetary mill (Pulverisette 6, Fritsch Gmbh, Weimar, Germany) using an agate jar and balls (10 balls of 30 mm in diameter). The resulting powder was then sieved over 30 min using a 40 µm sieve (Filtra, Barcelona, Spain), introducing 145 g of powder together with 200 g of Zirconia balls (1.5 mm diameter; Tosoh, Tokyo, Japan).
4.4. Molecular Weight Analysis
The molecular weight, LA/GA molar ratio and PLGA/PEG weight ratio of the copolymer were determined by means of proton nuclear magnetic resonance
1H NMR spectroscopy on a Bruker (Billerica, MA, USA) ADVANCE III HD 700 MHz instrument in deuterated chloroform (CDCl
3) solvent at 25 °C (
Supplementary Figure S1). The number-average molecular weight (
Mn) and polydispersity index (
Mw/
Mn) of the PLGA–PEG–PLGA copolymer were determined via gel permeation chromatography with the multi-angle light scattering method (GPC-MALS) (
Supplementary Figure S2). The instrumental setup included an Agilent HPLC 1100 Series instrument with a degasser, pump, autosampler, set of two PLgel 5 μm Mixed-C 300 × 7.5 mm columns (Agilent, Santa Clara, CA, USA) thermostated to 25 °C and a UV-VIS diode array detector connected to a DAWN HELEOS II multi-angle laser light scattering detector, a ViscoStar-II differential viscometer and Optilab T-rEX refractive index detectors (Wyatt Technology, Dernbach, Germany). Tetrahydrofuran was used as the mobile phase at a flow rate of 1 mL·min
−1.
4.5. Particle Size Analysis
The particle size distribution of the α-TCP powder was determined using a laser diffraction particle size analyser (LA-950, Horiba, Kyoto, Japan), and the specific surface area (SSA) of the powder was measured by means of nitrogen adsorption according to the 5-point Brunauer, Emmett and Teller (BET) method (Autosorb iQ-XR, Quantachrome Instruments, Boynton Beach, FL, USA).
4.6. Rheological Analysis
The rheological properties and tests (including the sol-gel transition, thixotropy, viscosity and dynamic time sweep tests) were conducted in steady mode using a controlled-stress rheometer (AR-G2, TA Instruments, New Castle, DE, USA).
4.6.1. Copolymer Thermo-Sensitivity Measurement
Cone-plate geometry with a diameter of 40 mm and a 2° angle was used for the temperature dependence measurement of the PLGA–PEG–PLGA triblock copolymer aqueous solution, which was transferred to the Peltier by means of a spatula. The working position was set with a geometry gap of 60 µm. A solvent trap filled with water was employed before the recording of the measurements so as to prevent the evaporation of the sample solvent. Experimental conditions were applied at a constant frequency of 1 rad s−1 and 1% strain. The temperature ramp was set up at from 15 °C to 60 °C and a rate of 0.5 °C per minute.
4.6.2. Copolymer/α-TCP Setting Kinetics
The time sweep test was conducted with plate-plate geometry (diameter 20 mm), at a frequency of 1 Hz, a points delay time of 10 s, a deformation of 0.01%; the measurement time was 5 h at a temperature of 23 °C for the first set of samples and 37 °C for the second set. The first set of samples included three types of paste with L/P ratios of 0.35; 0.5; 0.65 g·g−1 concerning which a liquid thermo-sensitive copolymer (with a concentration of 10 w/v% or 20 w/v%) or water (0 w/v%) were used as the liquid phase. The second set of samples at both 23 °C and 37 °C included pastes with an L/P ratio of 0.5 g·g−1 and differing in the amount of the polymer. The rheological measurements were taken 3 min following the mixing of the two components.
4.6.3. Copolymer/α-TCP Thixotropy and Viscosity
Parallel plate geometry with a diameter of 20 mm was used for measurement purposes with respect to the steady state rheological analysis. Samples of the prepared CPCs (0.16 mL) were transferred to the Peltier by means of a syringe. The AR-G2 rheometer (TA Instruments, New Castle, DE, USA) was set at the working position with a geometry gap of 500 µm. A solvent trap filled with water was employed so as to prevent solvent evaporation during time-sweep testing. The experimental conditions at 23 °C commenced at a shear rate of 1 s−1 which was maintained for 20 s followed by a rate increase to 100 1 s−1 which was also maintained at 20 s and a subsequent return to a shear rate of 1 s−1 according to the following scheme: . Each sample was measured at least three times.
4.7. Microstructure Observation
After 1 min of the mixing of α-TCP powder and a 20 w/v% polymer solution as the liquid phase, the cement paste with an L/P ratio of 0.5 g·g−1 was placed in cylindrical rubber moulds with an internal diameter of 6 mm and a height of 9 mm. The samples were then immersed in ultrapure water at 37 °C. At selected time-points (3, 6, 12 and 24 h, 3, 7, 14 and 28 days) one of the samples was extracted, rinsed with distilled water and quenched with liquid nitrogen so as to prevent a hydrolysis reaction and, finally, freeze-dried (EPSILON 2D-10, Martin Christ, Osterode am Harz, Germany). The morphology of the initial α-TCP powder and set cements was investigated employing a scanning electron microscope (SEM, Tescan Mira3, Brno, Czech Republic). All the observations were made in the secondary electron emission mode with a 10 kV acceleration voltage, beam intensity of 10 and a working distance of 15 mm. In order to ensure good resolution, the samples were coated with a 20 nm layer of gold/palladium.
4.8. Kinetics of the α-TCP Transformation to CDHA
The kinetics of the transformation from the original α-TCP to calcium-deficient hydroxyapatite (CDHA) of the CPC with the 20 w/v% polymer solution and L/P ratio of 0.5 g·g−1 was studied by means of Wide-Angle X-ray Scattering Analysis (WAXS, Rigaku Miniflex 6000, Tokyo, Japan). Following SEM observation, the remaining set cement samples were milled and analysed in Bragg-Brentano geometry, with an accelerating voltage of 40 kV and 15 mA, Cu-Kα radiation, with a D/teX detector, filter K(β) and a step width of 0.02° in a scanning range of 2° to 60° at laboratory temperature (21 ± 0.5 °C). The diffraction patterns of the analysed samples were collected and evaluated employing PDXL software.
4.9. Porosity Determination
Changes in the pore size distribution in the cement during setting were investigated by means of mercury intrusion porosimetry (PoreMaster 60, Quantachrome Instruments, Boynton Beach, FL, USA). The cement samples set over 10, 24 and 35 h, 3, 7 and 14 days by means of immersion in ultrapure water at 37 °C and, at the specified time-points, were withdrawn and freeze-dried (Martin Christ lyophilizator EPSILON 2D-10, Osterode am Harz, Germany).
4.10. Compression Testing
Compression testing was performed on the cement cylinders prepared as described in the “Microstructure observation” section using mechanical testing equipment (Zwick Roell 022, Ulm, Germany) with a load-cell of 1 kN, a crosshead speed of 1 mm min
−1 and a pre-load of 10 g. The compression testing of the samples at different reaction times and the samples immersed in the acidic solution for 4 and 8 h (see the “Accelerated degradation study”) was performed according to the ISO 13,314 standard [
40]. The ultimate compressive strength was calculated following the measurement of the ultimate compressive force. The mechanical data was collected in testXpert II software (Zwick Roell, Ulm, Germany) and each set consisted of 5 samples.
4.11. Accelerated Degradation Study
Accelerated degradation testing was conducted according to Escudero et al. [
24]. Cylindrical samples (
n = 5 for each time-point) with dimensions of 9 mm in diameter and 2 mm in height made from
α-TCP mixed with the 20
w/
v% copolymer (L/P ratio of 0.5 g·g
−1 set for 14 days) were dried at 120 °C overnight until a constant weight was achieved. Cylindrical samples with an acidic solution consisting of 0.01 M hydrochloric acid (HCl, Penta, Praha, Czech Republic) and 0.14 M sodium chloride (NaCl, Penta, Praha, Czech Republic) (pH = 2.2 at 37 °C) were placed in polypropylene tubes for 8 h. The solution was changed every hour and the pH values were checked at each time-point in the collected supernatants using an IKATRON THETA 90 electrode (IKA Werke, Staufen im Breisgau, Germany). The ratio of the solution volume and the surface area of the samples was kept constant (100 mL to 185 mm
2) for all the tests so as to maintain a constant pH during degradation testing.
Mass Loss and Calcium Ion Release
Following degradation, the samples were withdrawn from the acidic solution at specified times, washed three times in deionized water and dried at 120 °C until constant weight was achieved. The mass loss percentage was calculated according to the following Equation (2):
where
w0 is the initial dried weight of the sample and
wt is the dried weight of the sample following degradation. The calcium concentration of the solution was measured at each time-point using an HI4104 calcium ion selective electrode (HANNA Instruments, Woonsocket, RI, USA).
4.12. Cytocompatibility Testing
The cytocompatibility of the bone cements after 10 days of setting was tested using human mesenchymal stem cells (hMSCs; ScienCell, Carlsbad, CA, USA). Copolymer/α-TCP samples (L/P ratio of 0.5, 20 w/v% of copolymer) with a diameter of 6 mm were sterilized using ethylene oxide and seeded with hMSCs at a density of 2 × 104 per sample. The cells were cultured in αMEM supplemented with 10% foetal bovine serum (FBS) and 1% antibiotics (Penicillin/Streptomycin). Cells seeded on tissue culture plastic were used as a control group.
4.12.1. DNA Quantification
On days 1 and 3, the amount of cells on the scaffolds (cell proliferation) was determined by means of dsDNA quantification (Quant-iT™ dsDNA Assay Kit; Life Technologies, Carlsbad, CA, USA). The polymer/α-TCP samples seeded with cells were placed in 300 μL of cell lysis solution (0.2% v/v Triton X-100, 10 mM Tris (pH 7.0), and 1 mM EDTA) and processed through 3 freeze/thaw cycles (vortexed between each cycle). The samples were further processed according to the manufacturer’s instructions and measured using a multiplate fluorescence reader (Synergy HT, Winooski, VT, USA; λex = 485 nm, λem = 525 nm). The averaged dsDNA quantification values were determined after 24 and 72 h with respect to at least 5 independently prepared samples. The data were normalized to the values obtained after 24 h of cell culture.
4.12.2. Cell Distribution
Confocal microscopy was used for the visualisation of the distribution of cells on the cements. The copolymer/α-TCP samples were fixed with frozen methyl alcohol (−20 °C) for 10 min and subsequently incubated with 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3) (D273, Invitrogen, Molecular Probes, Eugene, OR, USA, 1 μg·mL−1 in phosphate buffered saline (PBS); pH 7.4) for 45 min at room temperature so as to visualize the cell membranes (in green). The samples were then incubated with propidium iodide (PI; 5 μg·mL−1 in PBS; Sigma-Aldrich, St. Louis, MO, USA) for 10 min so as to visualize the cell nuclei (in red). The samples were scanned using a ZEISS LSM 5 DUO confocal microscope, Oberkochen, Germany (PI: λex = 561 nm, λem = 630 − 700 nm; DiOC6 (3): λex = 488 nm, λem = 505 − 550 nm).
4.12.3. MTS Test
The MTS assay (CellTiter96® AQueous One Solution Cell Proliferation Assay, Promega, WI, USA) was used 24 and 70 h after seeding to determine the metabolic activity of the cells on the bone cements. Briefly, the scaffolds were transferred into new wells to prevent the cells adhered to the tissue culture plastic to misrepresent the measured data. Subsequently, 100 μL of fresh media and 20 μL of the MTS substrate were added to each well. After 2 h of incubation at 37 °C, 100 μL of the cultured solution was transferred to a new clean well. The absorbance of the media was detected at 490 nm using a multi-mode microplate reader Synergy HT (BioTek Instruments, Winooski, VT, USA). The background absorbance (690 nm) and the absorbance of the medium without cells were subtracted from the measured absorbance. The data were normalized to the values obtained after 24 h of cell culture.
4.13. Statistical Analysis
The mean values were calculated for the number of readings (n) from each experiment; the error bars refer to the standard deviation (SD). The cytocompatibility results were evaluated statistically using the One Way Analysis of Variance method and the Student-Newman-Keuls test (SigmaStat 12.0, Systat Software, San Jose, CA, USA). Statistical significance was accepted when p ≤ 0.05.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}