Arf GAPs as Regulators of the Actin Cytoskeleton—An Update


Abstract
:1. Introduction
2. Actin-Based Structures and Functions Affected by Arf GAPs
3. Arf and Rho Family GTP-Binding Proteins in Actin Remodeling
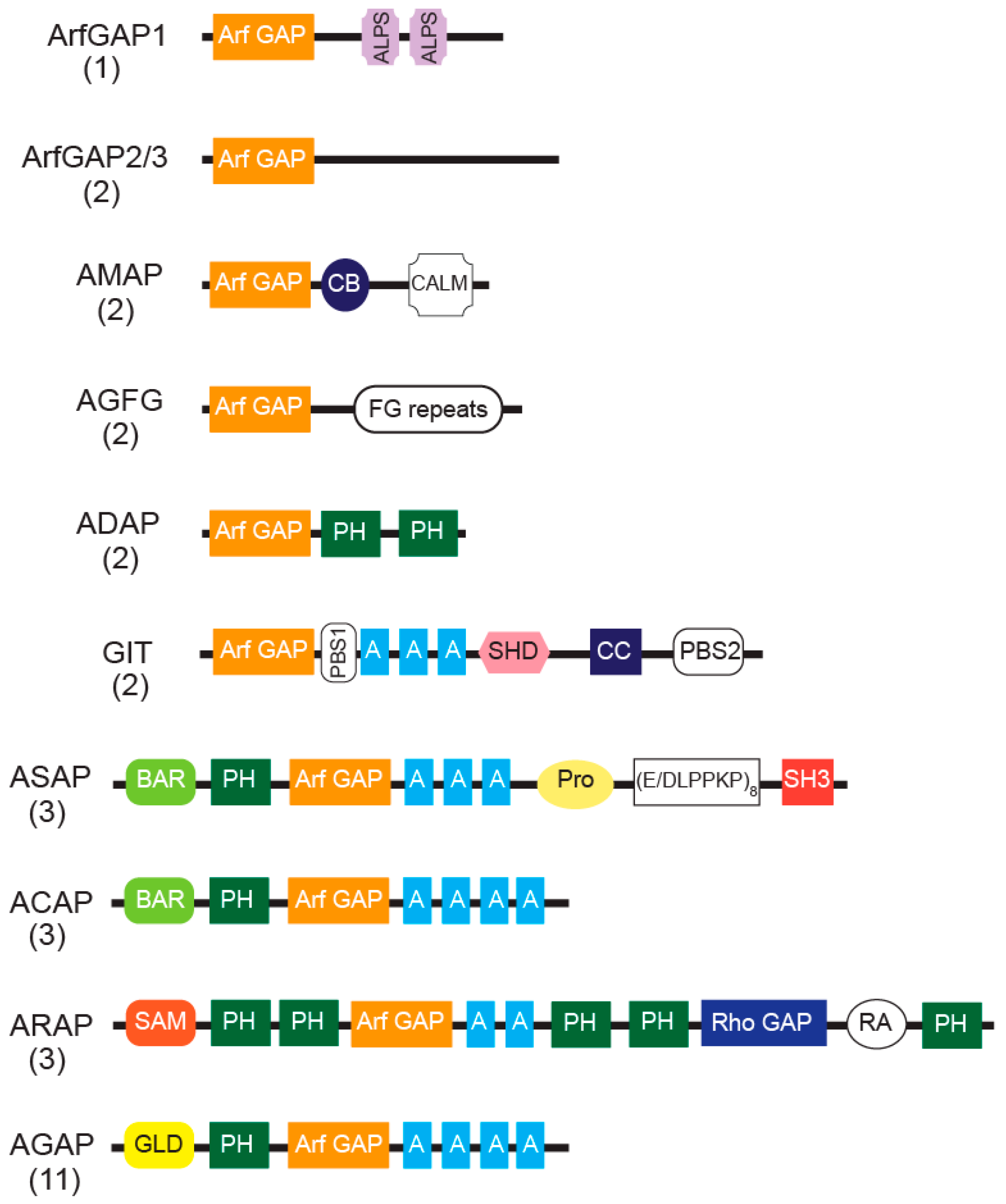
4. The Arf GAP Family
5. Arf GAPs that Regulate Circular Dorsal Ruffles (CDRs)
6. Arf GAPs that Regulate Podosomes and Invadopodia
7. Arf GAPs in Motility-Related Structures: Lamellipodia, Stress Fibers and Focal Adhesions
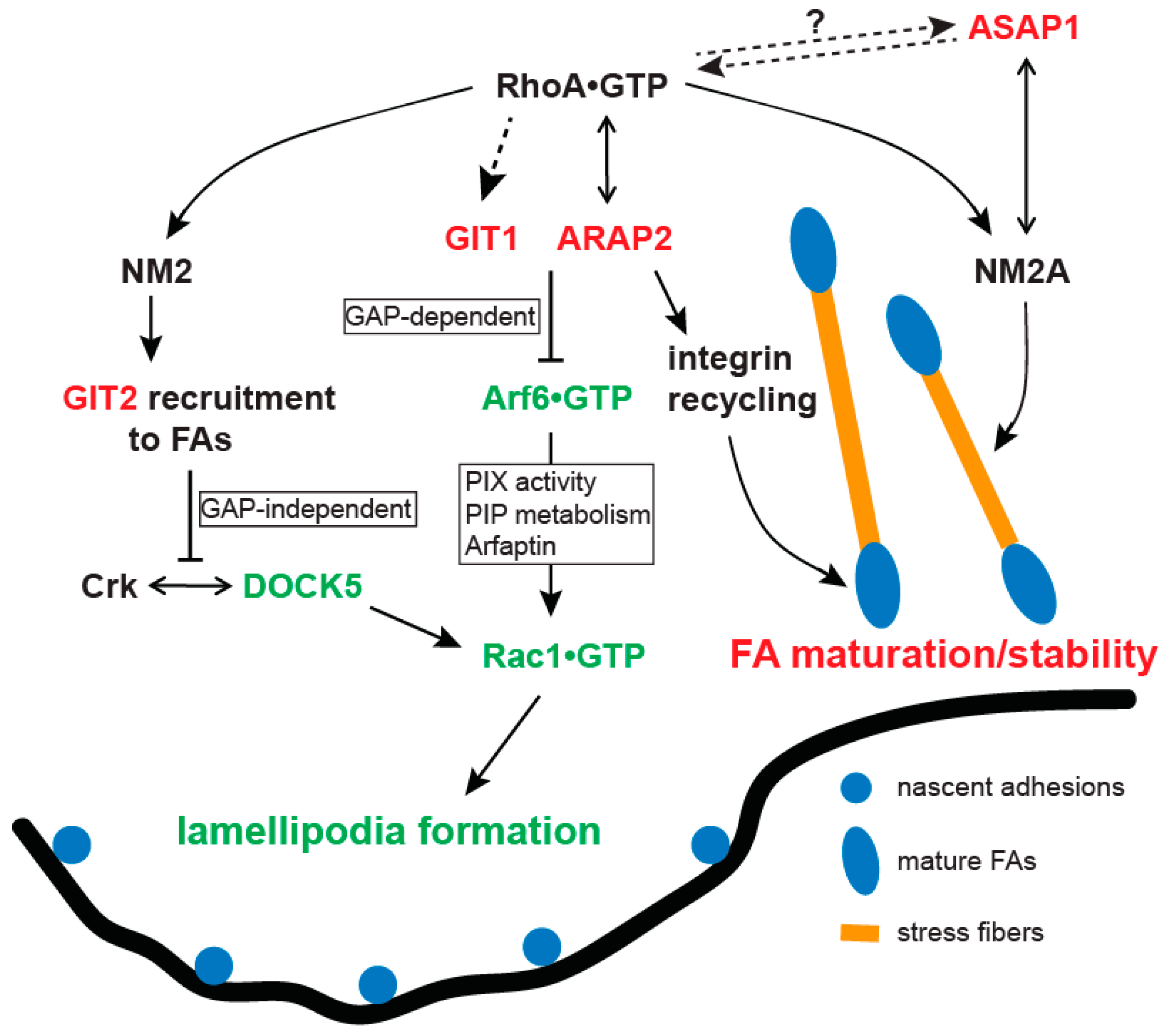
7.1. ASAP1
7.2. GIT1 and GIT2
7.3. ARAP2
7.4. ARAP1 and ARAP3
8. Arf GAPs in Engulfment of Pathogens and Apoptotic Cells
9. Arf GAPs that Regulate Neurite Outgrowth or Neuron Migration
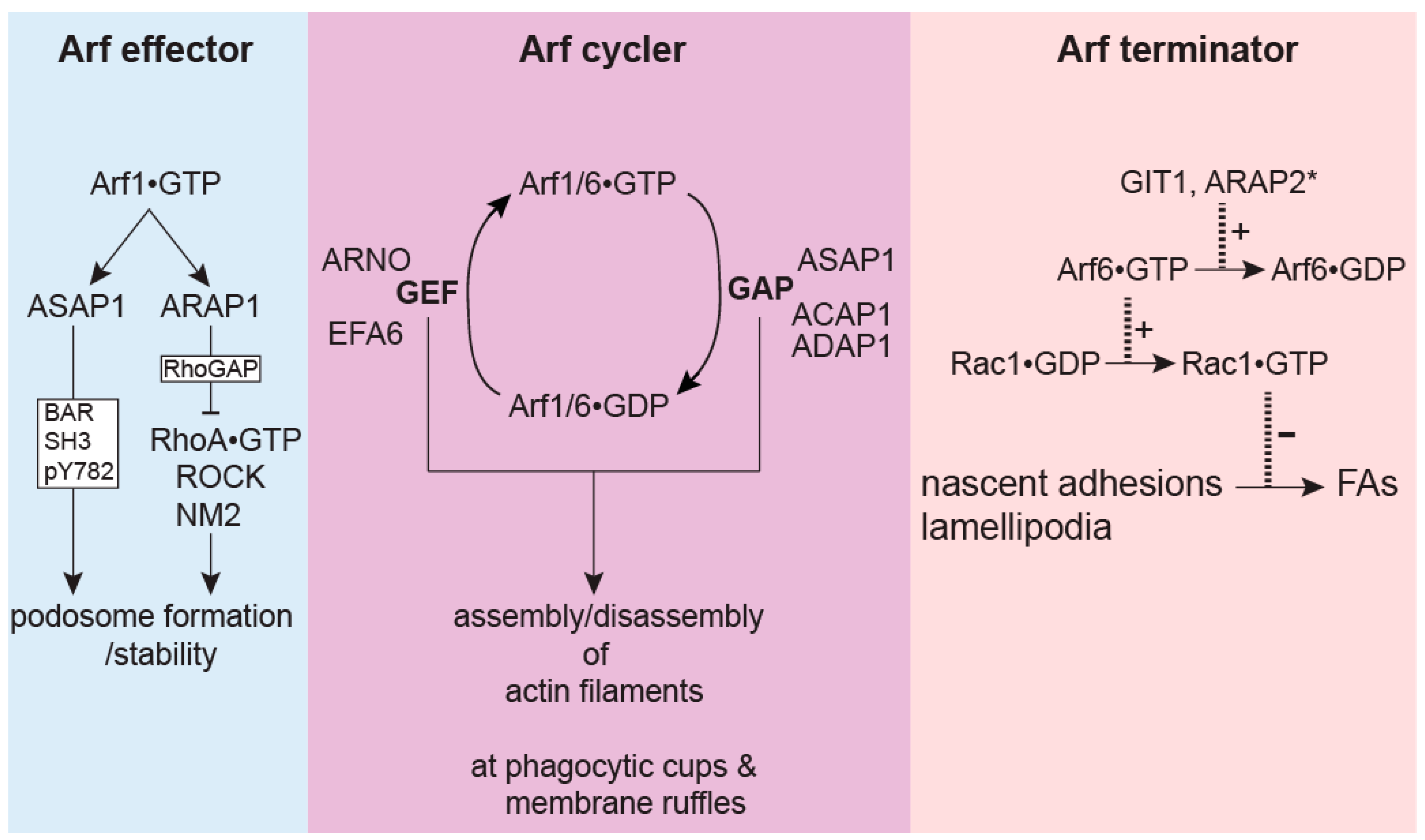
10. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALPS | Arf GAP1 lipid-packing sensor |
| Arf | ADP-ribosylation factor |
| Arf GAP | Arf GTPase-activating protein |
| Arls | Arf-like proeins |
| ARNO | ARF nucleotide-binding site opener |
| BAR | Bin/Amphiphysin/Rvs |
| CA | Constitutively active |
| CALM | CALM binding domain |
| CB | Clathrin-box |
| CC | Coiled-coil |
| CDR | Circular dorsal ruffles |
| Crk | Chicken tumor virus 10 regulator of kinase |
| DOCK | Dedicator of cytokinesis |
| DN | Dominant negative |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EFA6 | Exchange factor for Arf6 |
| ELMO | Engulfment and cell motility protein |
| ERK | Extracellular signal-regulated kinase |
| FA | Focal adhesion |
| F-actin | Filamentous actin/actin filaments |
| FAK | Focal adhesion kinase |
| GDI | Guanine nucleotide dissociation inhibitor |
| GEF | Guanine nucleotide exchange factor |
| GIT | G protein-coupled receptor kinase interactor |
| GLD | GTP-binding protein-like domain |
| GRK | G protein-coupled receptor kinase |
| Mtb | Mycobacterium tuberculosis |
| HGF | Hepatocyte growth factor |
| NGF | Nerve growth factor |
| NM2 | Nonmuscle myosin 2 |
| NM2A | Nonmuscle myosin 2A |
| N-WASP | neural Wiskott-Aldrich syndrome protein |
| PAK | p21-activated kinase |
| PBS | Paxillin binding site |
| PDGF | Platelet-derived growth factor |
| PH | pleckstrin homology domain |
| PIP | Phosphoinositides |
| PIX | PAK-interacting exchange factor |
| PI3K | Phosphoinositide 3-kinase |
| PI4P5K | Phosphatidylinositol 4-phosphate 5 kinase |
| PI(4,5)P2 | Phosphatidylinositol 4,5-bisphosphate |
| PI(3,4,5)P3 | Phosphatidylinositol 3,4,5-triphosphate |
| PLD | Phospholipase D |
| PMA | Phorbol 12-myristate 13-acetate |
| RA | Ras association motif |
| ROCK | Rho-associated kinase |
| SAM | Sterile α-motif |
| SARs | Secretion-associated and Ras-related proteins |
| SelK | Selenoprotein K |
| SHD | Spa-homology domain |
| SH3 | Src homology 3 domain |
| VEGF | Vascular endothelial growth factor |
| WAVE | WASP family veroprolin homologous protein |
| WRC | WAVE regulator complex |
References
- Bezanilla, M.; Gladfelter, A.S.; Kovar, D.R.; Lee, W.L. Cytoskeletal dynamics: A view from the membrane. J. Cell Biol. 2015, 209, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappalainen, P. Actin-binding proteins: The long road to understanding the dynamic landscape of cellular actin networks. Mol. Biol. Cell 2016, 27, 2519–2522. [Google Scholar] [CrossRef] [PubMed]
- Saarikangas, J.; Zhao, H.; Lappalainen, P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 2010, 90, 259–289. [Google Scholar] [CrossRef] [PubMed]
- Diz-Munoz, A.; Thurley, K.; Chintamen, S.; Altschuler, S.J.; Wu, L.F.; Fletcher, D.A.; Weiner, O.D. Membrane Tension Acts Through PLD2 and mTORC2 to Limit Actin Network Assembly During Neutrophil Migration. PLoS Biol. 2016, 14, e1002474. [Google Scholar] [CrossRef] [PubMed]
- Jarsch, I.K.; Daste, F.; Gallop, J.L. Membrane curvature in Cell.Biol.ogy: An integration of molecular mechanisms. J. Cell Biol. 2016, 214, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Egami, Y.; Taguchi, T.; Maekawa, M.; Arai, H.; Araki, N. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front. Physiol. 2014, 5, 374. [Google Scholar] [CrossRef] [PubMed]
- Kjos, I.; Vestre, K.; Guadagno, N.A.; Borg Distefano, M.; Progida, C. Rab and Arf proteins at the crossroad between membrane transport and cytoskeleton dynamics. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.R.; Casanova, J.E. Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol. 2008, 18, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Egami, Y.; Fujii, M.; Kawai, K.; Ishikawa, Y.; Fukuda, M.; Araki, N. Activation-Inactivation Cycling of Rab35 and ARF6 Is Required for Phagocytosis of Zymosan in RAW264 Macrophages. J. Immunol. Res. 2015, 2015, 429439. [Google Scholar] [CrossRef]
- Egami, Y.; Fukuda, M.; Araki, N. Rab35 regulates phagosome formation through recruitment of ACAP2 in macrophages during FcgammaR-mediated phagocytosis. J. Cell Sci. 2011, 124, 3557–3567. [Google Scholar] [CrossRef]
- Randazzo, P.A.; Inoue, H.; Bharti, S. Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell 2007, 99, 583–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, H.; Kondo, A.; Yoshimura, Y.; Mazaki, Y.; Sabe, H. PAG3/Papalpha/KIAA0400, a GTPase-activating protein for ADP-ribosylation factor (ARF), regulates ARF6 in Fcgamma receptor-mediated phagocytosis of macrophages. J. Exp. Med. 2001, 193, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Haase, M.; Cordes, N. Signalling via integrins: Implications for cell survival and anticancer strategies. Biochim. Biophys. Acta 2007, 1775, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Layseca, P.; Streuli, C.H. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C.H. Integrins and cell-fate determination. J. Cell Sci. 2009, 122, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Buccione, R.; Orth, J.D.; McNiven, M.A. Foot and mouth: Podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 2004, 5, 647–657. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The Cell Biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef]
- Hoon, J.L.; Wong, W.K.; Koh, C.G. Functions and regulation of circular dorsal ruffles. Mol. Cell Biol. 2012, 32, 4246–4257. [Google Scholar] [CrossRef]
- Itoh, T.; Hasegawa, J. Mechanistic insights into the regulation of circular dorsal ruffle formation. J. Biochem. 2013, 153, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, G.; Kay, R.R. Uses and abuses of macropinocytosis. J. Cell Sci. 2016, 129, 2697–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.M.; King, J.S. Drinking problems: Mechanisms of macropinosome formation and maturation. FEBS J. 2017, 284, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Song, O.R.; Queval, C.J.; Iantomasi, R.; Delorme, V.; Marion, S.; Veyron-Churlet, R.; Werkmeister, E.; Popoff, M.; Ricard, I.; Jouny, S.; et al. ArfGAP1 restricts Mycobacterium tuberculosis entry by controlling the actin cytoskeleton. EMBO Rep. 2018, 19, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.C.; Humphreys, D.; Brooks, A.B.; Hume, P.J.; Koronakis, V. The Arf GTPase-activating protein family is exploited by Salmonella enterica serovar Typhimurium to invade nonphagocytic host cells. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Jian, X.; Heissler, S.M.; Le, K.; Luo, R.; Jenkins, L.M.; Nagy, A.; Moss, J.; Sellers, J.R.; Randazzo, P.A. The Arf GTPase-activating Protein, ASAP1, Binds Nonmuscle Myosin 2A to Control Remodeling of the Actomyosin Network. J. Biol. Chem. 2016, 291, 7517–7526. [Google Scholar] [CrossRef]
- Randazzo, P.A.; Andrade, J.; Miura, K.; Brown, M.T.; Long, Y.Q.; Stauffer, S.; Roller, P.; Cooper, J.A. The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2000, 97, 4011–4016. [Google Scholar] [CrossRef] [Green Version]
- Bharti, S.; Inoue, H.; Bharti, K.; Hirsch, D.S.; Nie, Z.; Yoon, H.Y.; Artym, V.; Yamada, K.M.; Mueller, S.C.; Barr, V.A.; et al. Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol. Cell Biol. 2007, 27, 8271–8283. [Google Scholar] [CrossRef]
- Onodera, Y.; Hashimoto, S.; Hashimoto, A.; Morishige, M.; Mazaki, Y.; Yamada, A.; Ogawa, E.; Adachi, M.; Sakurai, T.; Manabe, T.; et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005, 24, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Loijens, J.C.; Martin, K.H.; Karginov, A.V.; Parsons, J.T. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell 2002, 13, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Wada, I.; Miura, K.; Okawa, K.; Kadoya, T.; Kato, T.; Nishihara, H.; Maeda, M.; Tanaka, S.; Nagashima, K.; et al. CrkL directs ASAP1 to peripheral focal adhesions. J. Biol. Chem. 2003, 278, 6456–6460. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.L.; Fredericks, G.J.; Huang, Z.; Fay, J.D.; Hoffmann, F.W.; Hoffmann, P.R. Selenoprotein K regulation of palmitoylation and calpain cleavage of ASAP2 is required for efficient FcgammaR-mediated phagocytosis. J. Leukoc. Biol. 2017, 101, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Ha, V.L.; Bharti, S.; Inoue, H.; Vass, W.C.; Campa, F.; Nie, Z.; de Gramont, A.; Ward, Y.; Randazzo, P.A. ASAP3 is a focal adhesion-associated Arf GAP that functions in cell migration and invasion. J. Biol. Chem. 2008, 283, 14915–14926. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.R.; Brown, F.D.; Nie, Z.; Miura, K.; Foroni, L.; Sun, J.; Hsu, V.W.; Donaldson, J.G.; Randazzo, P.A. ACAPs are arf6 GTPase-activating proteins that function in the cell periphery. J. Cell Biol. 2000, 151, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fukuda, M. Rab35 establishes the EHD1-association site by coordinating two distinct effectors during neurite outgrowth. J. Cell Sci. 2013, 126, 2424–2435. [Google Scholar] [CrossRef] [PubMed]
- Kanno, E.; Ishibashi, K.; Kobayashi, H.; Matsui, T.; Ohbayashi, N.; Fukuda, M. Comprehensive screening for novel rab-binding proteins by GST pull-down assay using 60 different mammalian Rabs. Traffic 2010, 11, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Etoh, K.; Fukuda, M. Structure-function analyses of the small GTPase Rab35 and its effector protein centaurin-beta2/ACAP2 during neurite outgrowth of PC12 cells. J. Biol. Chem. 2015, 290, 9064–9074. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Hongu, T.; Yamauchi, Y.; Funakoshi, Y.; Katagiri, N.; Ohbayashi, N.; Kanaho, Y. ACAP3 regulates neurite outgrowth through its GAP activity specific to Arf6 in mouse hippocampal neurons. Biochem. J. 2016, 473, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Kanaho, Y. ACAP3, the GTPase-activating protein specific to the small GTPase Arf6, regulates neuronal migration in the developing cerebral cortex. Biochem. Biophys. Res. Commun. 2017, 493, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Segeletz, S.; Danglot, L.; Galli, T.; Hoflack, B. ARAP1 Bridges Actin Dynamics and AP-3-Dependent Membrane Traffic in Bone-Digesting Osteoclasts. iScience 2018, 6, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, J.; Tsujita, K.; Takenawa, T.; Itoh, T. ARAP1 regulates the ring size of circular dorsal ruffles through Arf1 and Arf5. Mol. Biol. Cell 2012, 23, 2481–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, M.; Rikitake, Y.; Takahashi, M.; Nagamatsu, Y.; Yamauchi, Y.; Ogita, H.; Hirata, K.; Takai, Y. Regulation by afadin of cyclical activation and inactivation of Rap1, Rac1, and RhoA small G proteins at leading edges of moving NIH3T3 cells. J. Biol. Chem. 2009, 284, 24595–24609. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Jacques, K.M.; Stauffer, S.; Kubosaki, A.; Zhu, K.; Hirsch, D.S.; Resau, J.; Zheng, Y.; Randazzo, P.A. ARAP1: A point of convergence for Arf and Rho signaling. Mol. Cell 2002, 9, 109–119. [Google Scholar] [CrossRef]
- Chen, P.W.; Jian, X.; Yoon, H.Y.; Randazzo, P.A. ARAP2 signals through Arf6 and Rac1 to control focal adhesion morphology. J. Biol Chem. 2013, 288, 5849–5860. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Chen, P.W.; Kuo, J.C.; Jenkins, L.; Jian, X.; Waterman, C.M.; Randazzo, P.A. ARAP2 inhibits Akt independently of its effects on focal adhesions. Biol. Cell 2018, 110, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Miura, K.; Cuthbert, E.J.; Davis, K.K.; Ahvazi, B.; Casanova, J.E.; Randazzo, P.A. ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating-protein activity and binding to RhoA-GTP. J. Cell Sci. 2006, 119, 4650–4666. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.W.; Luo, R.; Jian, X.; Randazzo, P.A. The Arf6 GTPase-activating proteins ARAP2 and ACAP1 define distinct endosomal compartments that regulate integrin alpha5beta1 traffic. J. Biol. Chem. 2014, 289, 30237–30248. [Google Scholar] [CrossRef]
- Gavicherla, B.; Ritchey, L.; Gianfelice, A.; Kolokoltsov, A.A.; Davey, R.A.; Ireton, K. Critical role for the host GTPase-activating protein ARAP2 in InlB-mediated entry of Listeria monocytogenes. Infect. Immun. 2010, 78, 4532–4541. [Google Scholar] [CrossRef]
- Yu, C.H.; Rafiq, N.B.; Krishnasamy, A.; Hartman, K.L.; Jones, G.E.; Bershadsky, A.D.; Sheetz, M.P. Integrin-matrix clusters form podosome-like adhesions in the absence of traction forces. Cell Rep. 2013, 5, 1456–1468. [Google Scholar] [CrossRef]
- Yagi, R.; Tanaka, M.; Sasaki, K.; Kamata, R.; Nakanishi, Y.; Kanai, Y.; Sakai, R. ARAP3 inhibits peritoneal dissemination of scirrhous gastric carcinoma cells by regulating cell adhesion and invasion. Oncogene 2011, 30, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Krugmann, S.; Andrews, S.; Stephens, L.; Hawkins, P.T. ARAP3 is essential for formation of lamellipodia after growth factor stimulation. J. Cell Sci. 2006, 119, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, P.; Yin, G.; Smolock, E.M.; Zuscik, M.J.; Yan, C.; Berk, B.C. GPCR kinase 2 interacting protein 1 (GIT1) regulates osteoclast function and bone mass. J. Cell. Physiol. 2010, 225, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, S.K.; Cabrera, R.; Mao, S.P.H.; Christin, J.R.; Wu, B.; Guo, W.; Bravo-Cordero, J.J.; Condeelis, J.S.; Segall, J.E.; Hodgson, L. Rac3 regulates breast cancer invasion and metastasis by controlling adhesion and matrix degradation. J. Cell Biol. 2017, 216, 4331–4349. [Google Scholar] [CrossRef] [PubMed]
- Franchi, S.A.; Astro, V.; Macco, R.; Tonoli, D.; Barnier, J.V.; Botta, M.; de Curtis, I. Identification of a Protein Network Driving Neuritogenesis of MGE-Derived GABAergic Interneurons. Front. Cell. Neurosci. 2016, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Sowden, M.P.; Gerber, S.A.; Thomas, T.; Christie, C.K.; Mohan, A.; Yin, G.; Lord, E.M.; Berk, B.C.; Pang, J. G-protein-coupled receptor-2-interacting protein-1 is required for endothelial cell directional migration and tumor angiogenesis via cortactin-dependent lamellipodia formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Taba, Y.; Pang, J.; Yin, G.; Yan, C.; Berk, B.C. GIT1 mediates VEGF-induced podosome formation in endothelial cells: Critical role for PLCgamma. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.S.; Manser, E.; Loo, T.H.; Lim, L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 2000, 20, 6354–6363. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Tajima, M.; Kosako, H.; Nakaya, T.; Hashimoto, A.; Watari, K.; Nishihara, H.; Ohba, M.; Komiya, S.; Tani, N.; et al. GRK6 deficiency in mice causes autoimmune disease due to impaired apoptotic cell clearance. Nat. Commun. 2013, 4, 1532. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.R.; Adelstein, M.R.; Hansen, S.H. GIT2 represses Crk- and Rac1-regulated cell spreading and Cdc42-mediated focal adhesion turnover. EMBO J. 2006, 25, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.R.; Kollmann, C.P.; van Lidth de Jeude, J.F.; Thiagarajah, J.R.; Engelholm, L.H.; Frodin, M.; Hansen, S.H. The focal adhesion-associated proteins DOCK5 and GIT2 comprise a rheostat in control of epithelial invasion. Oncogene 2017, 36, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Tavano, S.; Filosa, G.; Gartner, A.; Pennucci, R.; Santambrogio, P.; Bachi, A.; Dotti, C.G.; de Curtis, I. Biochemical and functional characterisation of alphaPIX, a specific regulator of axonal and dendritic branching in hippocampal neurons. Biol. Cell 2012, 104, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Heckel, T.; Czupalla, C.; Expirto Santo, A.I.; Anitei, M.; Arantzazu Sanchez-Fernandez, M.; Mosch, K.; Krause, E.; Hoflack, B. Src-dependent repression of ARF6 is required to maintain podosome-rich sealing zones in bone-digesting osteoclasts. Proc. Natl. Acad. Sci. USA 2009, 106, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.; Stanley, K.T.; Stauffer, S.; Jacques, K.M.; Hirsch, D.S.; Takei, J.; Randazzo, P.A. AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J. Biol. Chem. 2002, 277, 48965–48975. [Google Scholar] [CrossRef]
- Dwane, S.; Durack, E.; O’Connor, R.; Kiely, P.A. RACK1 promotes neurite outgrowth by scaffolding AGAP2 to FAK. Cell. Signal. 2014, 26, 9–18. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, Y.; Kim, J.I.; Wang, Z.; Daaka, Y.; Nie, Z. Arf GTPase-activating protein AGAP2 regulates focal adhesion kinase activity and focal adhesion remodeling. J. Biol. Chem. 2009, 284, 13489–13496. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef]
- Kahn, R.A.; Cherfils, J.; Elias, M.; Lovering, R.C.; Munro, S.; Schurmann, A. Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J. Cell Biol. 2006, 172, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kelly, W.G.; Logsdon, J.M., Jr.; Schurko, A.M.; Harfe, B.D.; Hill-Harfe, K.L.; Kahn, R.A. Functional genomic analysis of the ADP-ribosylation factor family of GTPases: Phylogeny among diverse eukaryotes and function in C. elegans. FASEB J. 2004, 18, 1834–1850. [Google Scholar] [CrossRef]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small GTPases 2016, 7, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, T.; Paleotti, O.; Mironov, A.A.; Fraisier, V.; Stradal, T.E.; De Matteis, M.A.; Franco, M.; Chavrier, P. Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat. Cell Biol. 2005, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Hume, P.J.; Humphreys, D.; Liu, T.; Horning, O.; Jensen, O.N.; McGhie, E.J. WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc. Natl. Acad. Sci. USA 2011, 108, 14449–14454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, D.; Davidson, A.; Hume, P.J.; Koronakis, V. Salmonella virulence effector SopE and Host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe 2012, 11, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.; Liu, T.; Davidson, A.C.; Hume, P.J.; Koronakis, V. The Drosophila Arf1 homologue Arf79F is essential for lamellipodium formation. J. Cell Sci. 2012, 125, 5630–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Cohen, L.A.; Donaldson, J.G. Arf1 and Arf6 promote ventral actin structures formed by acute activation of protein kinase C and Src. Cytoskeleton 2014, 71, 380–394. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Ramirez, R.A.; Claing, A. ARF1 regulates adhesion of MDA-MB-231 invasive breast cancer cells through formation of focal adhesions. Cell. Signal. 2015, 27, 403–415. [Google Scholar] [CrossRef]
- Radhakrishna, H.; Klausner, R.D.; Donaldson, J.G. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J. Cell Biol. 1996, 134, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cox, D.; Tseng, C.C.; Donaldson, J.G.; Greenberg, S. A requirement for ARF6 in Fcgamma receptor-mediated phagocytosis in macrophages. J. Biol. Chem. 1998, 273, 19977–19981. [Google Scholar] [CrossRef]
- Marchesin, V.; Montagnac, G.; Chavrier, P. ARF6 promotes the formation of Rac1 and WAVE-dependent ventral F-actin rosettes in breast cancer cells in response to epidermal growth factor. PLoS ONE 2015, 10, e0121747. [Google Scholar] [CrossRef]
- Balasubramanian, N.; Scott, D.W.; Castle, J.D.; Casanova, J.E.; Schwartz, M.A. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 2007, 9, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmani, N.; Peglion, F.; Chavrier, P.; Etienne-Manneville, S. Cdc42 localization and cell polarity depend on membrane traffic. J. Cell Biol. 2010, 191, 1261–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishna, H.; Al-Awar, O.; Khachikian, Z.; Donaldson, J.G. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci. 1999, 112, 855–866. [Google Scholar] [PubMed]
- Koo, T.H.; Eipper, B.A.; Donaldson, J.G. Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Santy, L.C.; Ravichandran, K.S.; Casanova, J.E. The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr. Biol. 2005, 15, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Shi, B.; Jarzynka, M.J.; Yiin, J.J.; D’Souza-Schorey, C.; Cheng, S.Y. ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway. Cancer Res. 2009, 69, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.; Davidson, A.C.; Hume, P.J.; Makin, L.E.; Koronakis, V. Arf6 coordinates actin assembly through the WAVE complex, a mechanism usurped by Salmonella to invade host cells. Proc. Natl. Acad. Sci. USA 2013, 110, 16880–16885. [Google Scholar] [CrossRef] [Green Version]
- Kahn, R.A.; Bruford, E.; Inoue, H.; Logsdon, J.M., Jr.; Nie, Z.; Premont, R.T.; Randazzo, P.A.; Satake, M.; Theibert, A.B.; Zapp, M.L.; et al. Consensus nomenclature for the human ArfGAP domain-containing proteins. J. Cell Biol. 2008, 182, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Reed, C.E.; Sload, J.A.; Wordeman, L.; Randazzo, P.A.; Chen, P.W. Arf GAPs and molecular motors. Small GTPases 2017. [Google Scholar] [CrossRef] [PubMed]
- Corallino, S.; Malinverno, C.; Neumann, B.; Tischer, C.; Palamidessi, A.; Frittoli, E.; Panagiotakopoulou, M.; Disanza, A.; Malet-Engra, G.; Nastaly, P.; et al. A RAB35-p85/PI3K axis controls oscillatory apical protrusions required for efficient chemotactic migration. Nat. Commun. 2018, 9, 1475. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fukuda, M. Rab35 regulates Arf6 activity through centaurin-beta2 (ACAP2) during neurite outgrowth. J. Cell Sci. 2012, 125, 2235–2243. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, J.; Tokuda, E.; Tenno, T.; Tsujita, K.; Sawai, H.; Hiroaki, H.; Takenawa, T.; Itoh, T. SH3YL1 regulates dorsal ruffle formation by a novel phosphoinositide-binding domain. J. Cell Biol. 2011, 193, 901–916. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, N.B.; Lieu, Z.Z.; Jiang, T.; Yu, C.H.; Matsudaira, P.; Jones, G.E.; Bershadsky, A.D. Podosome assembly is controlled by the GTPase ARF1 and its nucleotide exchange factor ARNO. J. Cell Biol. 2017, 216, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.; Thrasher, A.J.; Blundell, M.P.; Machesky, L.; Jones, G.E. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood 2001, 98, 1142–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, Y.; Randazzo, P.A. GEFH1 binds ASAP1 and regulates podosome formation. Biochem. Biophys. Res. Commun. 2011, 406, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali, T.; Girald-Berlingeri, S.; Randazzo, P.A.; Chen, P.W. Arf GAPs: A family of proteins with disparate functions that converge on a common structure, the integrin adhesion complex. Small GTPases 2017. [Google Scholar] [CrossRef]
- Zhou, W.; Li, X.; Premont, R.T. Expanding functions of GIT Arf GTPase-activating proteins, PIX Rho guanine nucleotide exchange factors and GIT-PIX complexes. J. Cell Sci. 2016, 129, 1963–1974. [Google Scholar] [CrossRef]
- Chang, J.S.; Su, C.Y.; Yu, W.H.; Lee, W.J.; Liu, Y.P.; Lai, T.C.; Jan, Y.H.; Yang, Y.F.; Shen, C.N.; Shew, J.Y.; et al. GIT1 promotes lung cancer cell metastasis through modulating Rac1/Cdc42 activity and is associated with poor prognosis. Oncotarget 2015, 6, 36278–36291. [Google Scholar] [CrossRef]
- Ammer, A.G.; Weed, S.A. Cortactin branches out: Roles in regulating protrusive actin dynamics. Cell Motil. Cytoskel. 2008, 65, 687–707. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, A.; Yin, X.; Dong, S.; Luo, F.; Dou, C.; Lan, X.; Xie, Z.; Hou, T.; Xu, J.; et al. Mesenchymal stem cells promote endothelial progenitor cell migration, vascularization, and bone repair in tissue-engineered constructs via activating CXCR2-Src-PKL/Vav2-Rac1. FASEB J. 2018, 32, 2197–2211. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, N.; Kiosses, W.B.; Han, J.; Ginsberg, M.H. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat. Cell Biol. 2005, 7, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Manabe, R.; Kovalenko, M.; Webb, D.J.; Horwitz, A.R. GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 2002, 115, 1497–1510. [Google Scholar] [PubMed]
- Honda, A.; Nogami, M.; Yokozeki, T.; Yamazaki, M.; Nakamura, H.; Watanabe, H.; Kawamoto, K.; Nakayama, K.; Morris, A.J.; Frohman, M.A.; et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 1999, 99, 521–532. [Google Scholar] [CrossRef]
- Tarricone, C.; Xiao, B.; Justin, N.; Walker, P.A.; Rittinger, K.; Gamblin, S.J.; Smerdon, S.J. The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 2001, 411, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.C.; Han, X.; Hsiao, C.T.; Yates, J.R., III; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Laurin, M.; Cote, J.F. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014, 28, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krugmann, S.; Williams, R.; Stephens, L.; Hawkins, P.T. ARAP3 is a PI3K- and rap-regulated GAP for RhoA. Curr. Biol. 2004, 14, 1380–1384. [Google Scholar] [CrossRef]
- Krugmann, S.; Anderson, K.E.; Ridley, S.H.; Risso, N.; McGregor, A.; Coadwell, J.; Davidson, K.; Eguinoa, A.; Ellson, C.D.; Lipp, P.; et al. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol. Cell 2002, 9, 95–108. [Google Scholar] [CrossRef]
- Stacey, T.I.; Nie, Z.; Stewart, A.; Najdovska, M.; Hall, N.E.; He, H.; Randazzo, P.A.; Lock, P. ARAP3 is transiently tyrosine phosphorylated in cells attaching to fibronectin and inhibits cell spreading in a RhoGAP-dependent manner. J. Cell Sci. 2004, 117, 6071–6084. [Google Scholar] [CrossRef] [Green Version]
- Kondo, A.; Hashimoto, S.; Yano, H.; Nagayama, K.; Mazaki, Y.; Sabe, H. A new paxillin-binding protein, PAG3/Papalpha/KIAA0400, bearing an ADP-ribosylation factor GTPase-activating protein activity, is involved in paxillin recruitment to focal adhesions and cell migration. Mol. Biol. Cell 2000, 11, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Andreev, J.; Simon, J.P.; Sabatini, D.D.; Kam, J.; Plowman, G.; Randazzo, P.A.; Schlessinger, J. Identification of a new Pyk2 target protein with Arf-GAP activity. Mol. Cell Biol. 1999, 19, 2338–2350. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 2000, 103, 501–510. [Google Scholar] [CrossRef]
- Singh, M.K.; Gao, H.; Sun, W.; Song, Z.; Schmalzigaug, R.; Premont, R.T.; Zhang, Q. Structure-activity relationship studies of QS11, a small molecule Wnt synergistic agonist. Bioorg. Med. Chem. Lett. 2015, 25, 4838–4842. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Gomez, T.M.; Letourneau, P.C. Actin dynamics in growth cone motility and navigation. J. Neurochem. 2014, 129, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Santy, L.C. Characterization of a fast cycling ADP-ribosylation factor 6 mutant. J. Biol. Chem. 2002, 277, 40185–40188. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Fukaya, M.; Hayashi, K.; Kawauchi, T.; Nakajima, K.; Sakagami, H. ADP Ribosylation Factor 6 Regulates Neuronal Migration in the Developing Cerebral Cortex through FIP3/Arfophilin-1-dependent Endosomal Trafficking of N-cadherin. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Campa, F.; Yoon, H.Y.; Ha, V.L.; Szentpetery, Z.; Balla, T.; Randazzo, P.A. A PH domain in the Arf GTPase-activating protein (GAP) ARAP1 binds phosphatidylinositol 3,4,5-trisphosphate and regulates Arf GAP activity independently of recruitment to the plasma membranes. J. Biol. Chem. 2009, 284, 28069–28083. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Tang, W.K.; Zhai, P.; Roy, N.S.; Luo, R.; Gruschus, J.M.; Yohe, M.E.; Chen, P.W.; Li, Y.; Byrd, R.A.; Xia, D.; Randazzo, P.A. Molecular Basis for Cooperative Binding of Anionic Phospholipids to the PH Domain of the Arf GAP ASAP1. Structure 2015, 23, 1977–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedergang, F.; Colucci-Guyon, E.; Dubois, T.; Raposo, G.; Chavrier, P. ADP ribosylation factor 6 is activated and controls membrane delivery during phagocytosis in macrophages. J. Cell Biol. 2003, 161, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Arf GAP | Domains Required for Function | Actin Structures Affected | Function | Reference |
|---|---|---|---|---|
| ArfGAP1 | Arf GAP | Stress fibers | Limits stress fiber formation to restrict mycobacterial entry | [25] |
| ADAP1 | Arf GAP | Actin-based membrane ruffles | Facilitates Arf6 GTP/GDP cycles and actin remodeling necessary for Salmonella invasion | [26] |
| ASAP1 | Arf GAP, BAR | CDRs | Inhibits CDR formation through NM2A/GAP activity | [27,28] |
| SH3, Src-mediated phosphorylation, BAR, (Arf GAP-independent) | Podosomes and Invadopodia | Promotes podosome formation in fibroblasts and invadopodia in cancer cells | [29] | |
| Arf GAP | N.D. | Promotes migration and invasion of MDA-MB-231 cells | [30] | |
| N.D. (Not-determined) | Stress fibers, focal adhesions (FAs) | Increases mature FAs and assembly of stress fibers | [27] | |
| Arf GAP (partly) | Inhibits cell spreading in REF52 cells | [31] | ||
| SH3, Proline-rich | Targets to FAs | [31,32] | ||
| Arf GAP | Actin-based membrane ruffles | Facilitates Arf1 GTP/GDP cycles and actin remodeling necessary for Salmonella invasion | [26] | |
| ASAP2 | Arf GAP | F-actin structures at phagocytic cup | Regulates FcγR-mediated phagocytosis, potentially promotes by facilitating Arf6 GTP/GDP cycles | [12] |
| BAR | Phagocytic cup association | Regulates FcγR-mediated phagocytosis under control of Selk | [33] | |
| ASAP3 | N.D. | Stress fibers | Facilitates stress fiber formation, migration and invasion of MDA-MB-231 cells | [34] |
| ACAP1 | Arf GAP (partly) | CDRs | Inhibits CDR formation through GAP activity | [35] |
| Arf GAP | Actin-based membrane ruffles | Facilitates Arf6 GTP/GDP cycles and actin remodeling necessary for Salmonella invasion | [26] | |
| ACAP2 | Arf GAP (partly) | CDRs | Inhibits CDR formation through GAP activity | [35] |
| Arf GAP | Phagocytosis/phagocytic cups | Regulates FcyR- or zymosan-induced phagocytosis by facilitating Arf6 GTP/GDP cycles under control of Rab35 GTP/GDP cycles | [9,10] | |
| Ank | Rab35•GTP-dependent recruitment to phagocytic cups | Regulates FcγR-mediated phagocytosis under control of Rab35 GTP/GDP | [10] | |
| Arf GAP | Neurite outgrowth in PC12 cells | [36,37] | ||
| Ank | Rab35•GTP-dependent recruitment to plasma membrane | Neurite outgrowth in PC12 cells | [37,38] | |
| ACAP3 | Arf GAP | Uni/bipolar morphology of migrating neurons | Promotes neurite outgrowth by facilitating Arf6 GTP/GDP cycles in hippocampal neurons | [39] |
| Arf GAP | N.D. | Promotes neuron migration in developing cerebral cortex | [40] | |
| ARAP1 | PH3-PH4-Rho GAP-RA-PH5 (Arf GAP-independent) | Podosomes/sealing zones | Promotes dynamics and formation of podosome belt to aid bone resorption in osteoclasts | [41] |
| Arf GAP | CDRs | Regulates CDR ring size and macropinocytosis in NIH3T3 fibroblasts | [42] | |
| PH3-PH4-Rho GAP-RA-PH5 | CDR-targeting but not effect on CDRs | [42] | ||
| Rho GAP, RA (mediates Rap•GTP binding) | Lamellipodia, focal complexes, stress fibers | Promotes the formation of leading edge structures in migrating NIH 3T3 fibroblasts | [43] | |
| Arf GAP | Filopodia | Promotes filopodia formation in NIH 3T3 and HEK293T cells by activating Cdc42 activation and controlling its distribution | [44] | |
| Rho GAP | Stress fibers | Moderates stress fibers in NIH3T3 cells | [44] | |
| ARAP2 | Arf GAP, Rho GAP (RhoA•GTP binding, not GAP activity) | Focal adhesions, stress fibers | Promotes focal adhesion growth and stress fiber formation in HeLa, MDA-MB-231, and U118 glioblastoma cells | [45,46,47] |
| Arf GAP | Focal adhesions | Controls integrin β1 recycling in HeLa cells at APPL1 endosomes | [48] | |
| Arf GAP, Rho GAP (RhoA•GTP binding, not GAP activity) | F-actin structures around Listeria InB-coated beads | Promotes Listeria engulfment and F-actin enrichment around InB-coated beads | [49] | |
| ARAP3 | Rho GAP | Podosome-like adhesions | Mediates the response to a lack of traction forces in nontransformed fibroblasts on fluid surfaces | [50] |
| Rho GAP | Filopodia, lamellipodia | Inhibits motility, invasion and adhesion of scirrhous gastric carcinoma cells | [51] | |
| N.D. | Lamellipodia, focal adhesions, stress fibers | Mediates the response of PAE cells to growth factor simulation | [52] | |
| GIT1 | N.D. | Podosomes | Promotes bone resorption activity in osteoclasts | [53] |
| Arf GAP | Invadopodia | Facilitates the regulation of ECM degradation by Rac3 in MTLn3 cells | [54] | |
| SHD, PBS2 | Growth cone | Regulates neurite extension and branching | [55] | |
| SHD | Lamellipodia | Promotes directional migration of endothelial cells towards VEGF | [56] | |
| N.D. | Podosomes | Mediates the response to VEGF and promotes ECM degradation and migration in endothelial cells | [57] | |
| SHD | Focal complexes/adhesions | Promotes focal complex disassembly and motility in fibroblasts and epithelial cells | [58] | |
| CC | N.D. | Enhances GRK6-mediated phagocytosis of apoptotic cells by inhibiting Rac1 | [59] | |
| GIT2 | N.D. | Lamellipodia, focal adhesions | Inhibits lamellipodia formation, stabilizes focal adhesions and attenuates invasion of mammary epithelial cells | [60,61] |
| N.D. | Filopodia | Induces filopodia in growth cones, promotes neurite branching in hippocampal neurons | [62] | |
| N.D. | Podosomes/sealing zones | Promotes podosome formation | [63] | |
| AGAP1 | Arf GAP (partly) | CDRs, stress fibers | Inhibits formation of CDRs and stress fibers | [64] |
| AGAP2 | GLD (partly), (Arf GAP-independent) | Focal adhesions | Disassembly of FAs in HEK293, U87 and PC12 cells; promotes neurite outgrowth in PC12 cells | [65,66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanna, C.E.; Goss, L.B.; Ludwig, C.G.; Chen, P.-W. Arf GAPs as Regulators of the Actin Cytoskeleton—An Update. Int. J. Mol. Sci. 2019, 20, 442. https://doi.org/10.3390/ijms20020442
Tanna CE, Goss LB, Ludwig CG, Chen P-W. Arf GAPs as Regulators of the Actin Cytoskeleton—An Update. International Journal of Molecular Sciences. 2019; 20(2):442. https://doi.org/10.3390/ijms20020442
Chicago/Turabian StyleTanna, Christine E., Louisa B. Goss, Calvin G. Ludwig, and Pei-Wen Chen. 2019. "Arf GAPs as Regulators of the Actin Cytoskeleton—An Update" International Journal of Molecular Sciences 20, no. 2: 442. https://doi.org/10.3390/ijms20020442
APA StyleTanna, C. E., Goss, L. B., Ludwig, C. G., & Chen, P. -W. (2019). Arf GAPs as Regulators of the Actin Cytoskeleton—An Update. International Journal of Molecular Sciences, 20(2), 442. https://doi.org/10.3390/ijms20020442