Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis


Abstract
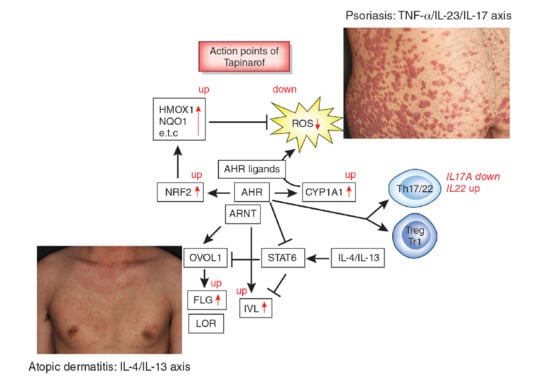
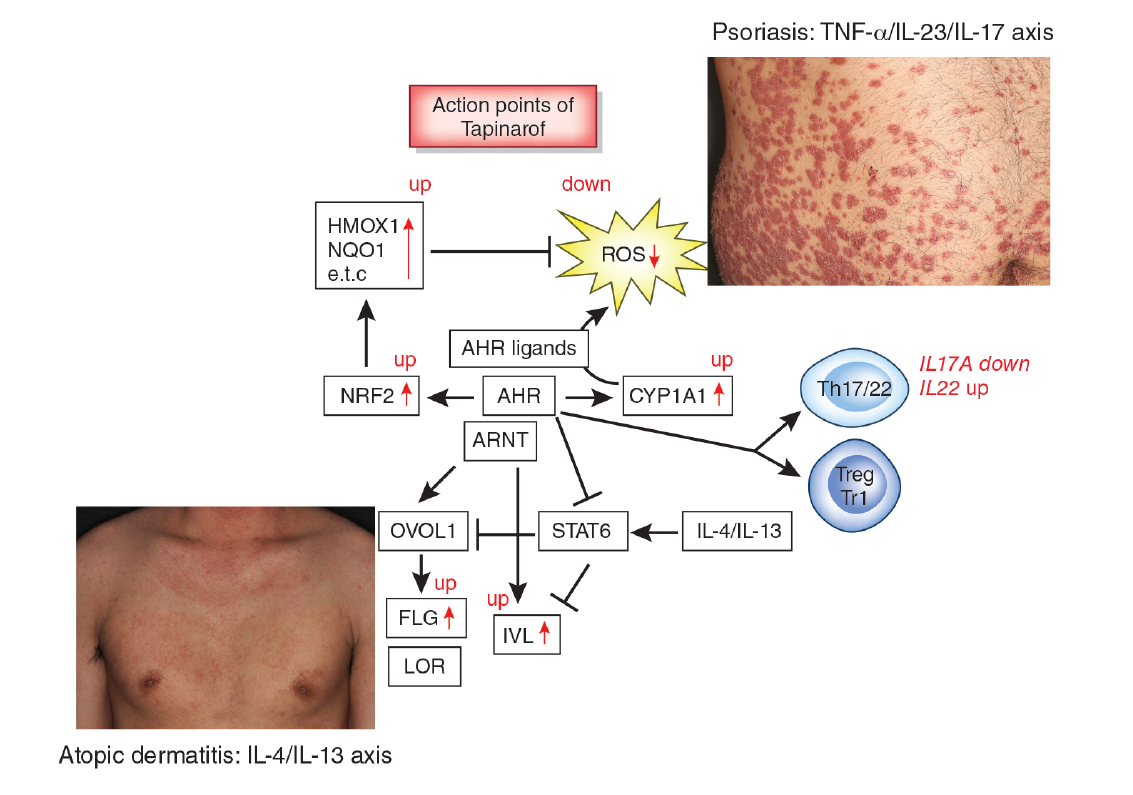
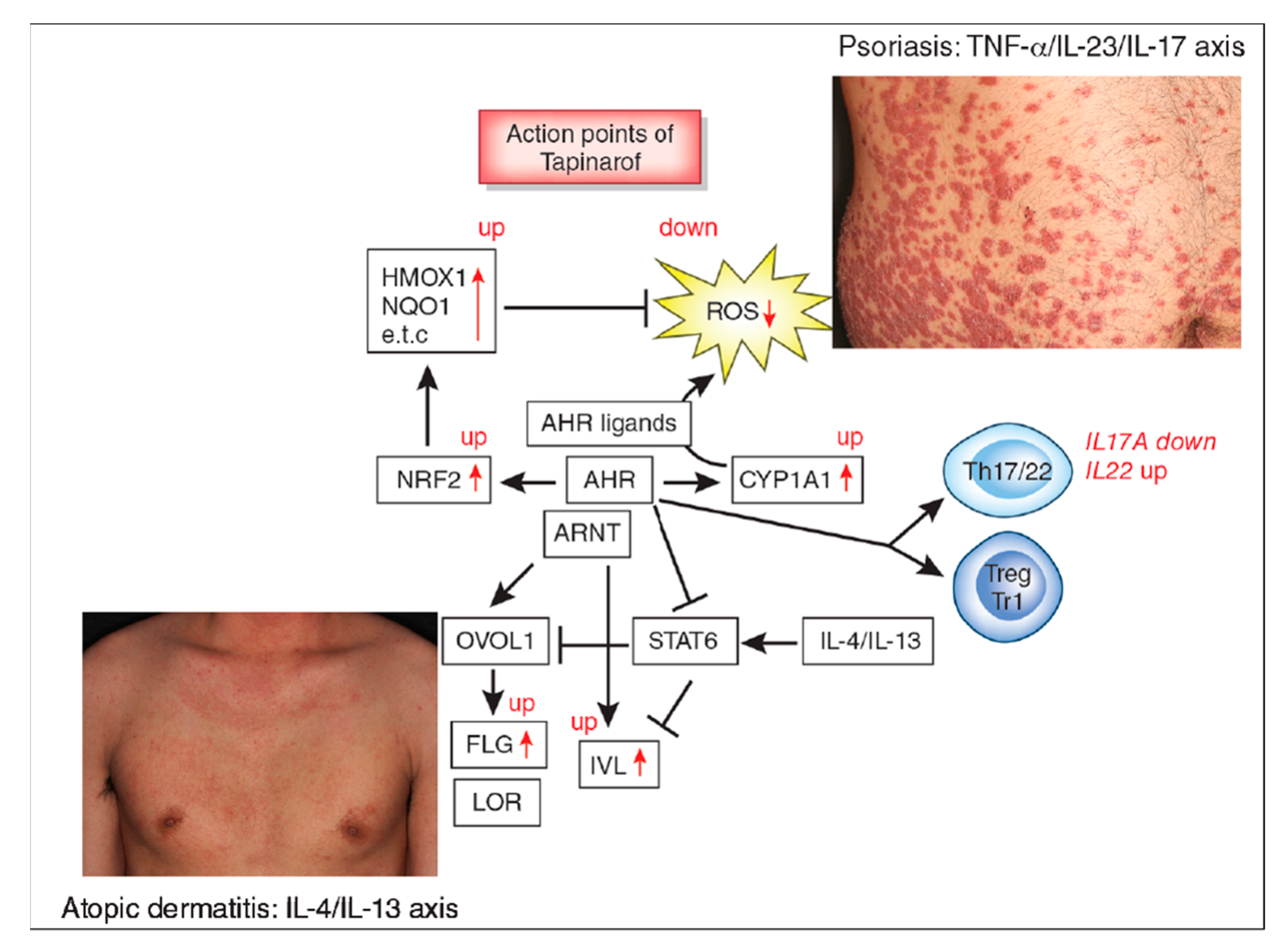
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. AHR Signaling and Modulation of Oxidative and Antioxidative Balance
3. AHR and Epidermal Terminal Differentiation
4. AHR and Immune Modulation
5. AHR and Atopic Dermatitis
6. AHR and Psoriasis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Furue, K.; Mitoma, C.; Tsuji, G.; Furue, M. Protective role of peroxisome proliferator-activated receptor α agonists in skin barrier and inflammation. Immunobiology 2018, 223, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Fuyuno, Y.; Mitoma, C.; Uchi, H.; Tsuji, G. Therapeutic agents with AHR inhibiting and NRF2 activating activity for managing chloracne. Antioxidants (Basel) 2018, 7, 90. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Antioxidative phytochemicals accelerate epidermal terminal differentiation via the AHR-OVOL1 pathway: Implications for atopic dermatitis. Acta Derm. Venereol. 2018, 98, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic metabolism, disposition, and regulation by receptors: From biochemical phenomenon to predictors of major toxicities. Toxicol. Sci. 2011, 120, S49–S75. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Bargen, I.; Weighardt, H.; Haarmann-Stemmann, T.; Krutmann, J. Functions of the aryl 1002 hydrocarbon receptor in the skin. Semin. Immunopathol. 2013, 35, 677–691. [Google Scholar] [CrossRef]
- Furue, M.; Takahara, M.; Nakahara, T.; Uchi, H. Role of AhR/ARNT system in skin homeostasis. Arch. Dermatol. Res. 2014, 306, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Fritsche, E.; Schäfer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hübenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef]
- Rannug, A.; Rannug, U.; Rosenkranz, H.S.; Winqvist, L.; Westerholm, R.; Agurell, E.; Grafström, A.K. Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J. Biol. Chem. 1987, 262, 15422–15427. [Google Scholar]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for healthy skin: The emerging role of aryl hydrocarbon receptors and nuclear factor-erythroid 2-related factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Magiatis, P.; Pappas, P.; Gaitanis, G.; Mexia, N.; Melliou, E.; Galanou, M.; Vlachos, C.; Stathopoulou, K.; Skaltsounis, A.L.; Marselos, M.; et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J. Investig. Dermatol. 2013, 133, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Galactomyces fermentation filtrate prevents T helper 2-mediated reduction of filaggrin in an aryl hydrocarbon receptor-dependent manner. Clin. Exp. Dermatol. 2015, 40, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Cynaropicrin attenuates UVB-induced oxidative stress via the AhR-Nrf2-Nqo1 pathway. Toxicol. Lett. 2015, 234, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Uchi, H.; Takahara, M.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Antioxidant soybean tar Glyteer rescues T-helper-mediated downregulation of filaggrin expression via aryl hydrocarbon receptor. J. Dermatol. 2015, 42, 171–180. [Google Scholar] [CrossRef]
- Van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two Phase 3 Trials of dupilumab versus placebo in atopic dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of atopic dermatitis: Current paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar]
- Furue, K.; Ito, T.; Furue, M. Differential efficacy of biologic treatments targeting the TNF-α/IL-23/IL-17 axis in psoriasis and psoriatic arthritis. Cytokine 2018, 111, 182–188. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Furue, M. Psoriasis and the TNF/IL23/IL17 axis. G. Ital. Dermatol. Venereol. 2019, 154, 418–424. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Robbins, K.; Bissonnette, R.; Maeda-Chubachi, T.; Ye, L.; Peppers, J.; Gallagher, K.; Kraus, J.E. Phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of plaque psoriasis. J. Am. Acad. Dermatol. 2019, 80, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, A.; Sundström, S.; Poellinger, L.; Pongratz, I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–2607. [Google Scholar] [CrossRef] [PubMed]
- Lees, M.J.; Peet, D.J.; Whitelaw, M.L. Defining the role for XAP2 in stabilization of the dioxin receptor. J. Biol. Chem. 2003, 278, 35878–35888. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001, 174, 103–113. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef]
- Esser, C. The aryl hydrocarbon receptor in immunity: Tools and potential. Methods Mol. Biol. 2016, 1371, 239–257. [Google Scholar]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Kopf, P.G.; Walker, M.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A. Toxicol. Appl. Pharmacol. 2010, 245, 91–99. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.M.; Höller, D.; Schiffer, R.; Frankenberg, S.; Neis, M.; Merk, H.F.; Jugert, F.K. Expression of multiple cytochrome p450 enzymes and multidrug resistance-associated transport proteins in human skin keratinocytes. J. Investig. Dermatol. 2001, 116, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Inui, H.; Itoh, T.; Yamamoto, K.; Ikushiro, S.; Sakaki, T. Mammalian cytochrome P450-dependent metabolism of polychlorinated dibenzo-p-dioxins and coplanar polychlorinated biphenyls. Int. J. Mol. Sci. 2014, 15, 14044–14057. [Google Scholar] [CrossRef] [PubMed]
- Anandasadagopan, S.K.; Singh, N.M.; Raza, H.; Bansal, S.; Selvaraj, V.; Singh, S.; Chowdhury, A.R.; Leu, N.A.; Avadhani, N.G. β-Naphthoflavone-induced mitochondrial respiratory damage in Cyp1 knockout mouse and in cell culture systems: Attenuation by resveratrol treatment. Oxid. Med. Cell Longev. 2017, 2017, 5213186. [Google Scholar] [CrossRef]
- Tanaka, Y.; Uchi, H.; Hashimoto-Hachiya, A.; Furue, M. Tryptophan photoproduct FICZ upregulates IL1A, IL1B, and IL6 expression via oxidative stress in keratinocytes. Oxid. Med. Cell. Longev. 2018, 2018, 9298052. [Google Scholar] [CrossRef]
- Tsuji, G.; Takahara, M.; Uchi, H.; Takeuchi, S.; Mitoma, C.; Moroi, Y.; Furue, M. An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J. Dermatol. Sci. 2011, 62, 42–49. [Google Scholar] [CrossRef]
- Nakahara, T.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Uchi, H.; Yan, X.; Hachisuka, J.; Chiba, T.; Esaki, H.; et al. Antioxidant Opuntia ficus-indica extract activates AHR-NRF2 signaling and upregulates filaggrin and loricrin expression in human keratinocytes. J. Med. Food 2015, 18, 1143–1149. [Google Scholar] [CrossRef]
- Doi, K.; Mitoma, C.; Nakahara, T.; Uchi, H.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, M.; Furue, M. Antioxidant Houttuynia cordata extract upregulates filaggrin expression in an aryl hydrocarbon-dependent manner. Fukuoka Igaku Zasshi 2014, 105, 205–213. [Google Scholar]
- Tsuji, G.; Takahara, M.; Uchi, H.; Matsuda, T.; Chiba, T.; Takeuchi, S.; Yasukawa, F.; Moroi, Y.; Furue, M. Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: The basis of its anti-inflammatory effect. J. Investig. Dermatol. 2012, 132, 59–68. [Google Scholar] [CrossRef]
- Yeager, R.L.; Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 2009, 111, 238–246. [Google Scholar] [CrossRef]
- Wang, K.; Lv, Q.; Miao, Y.M.; Qiao, S.M.; Dai, Y.; Wei, Z.F. Cardamonin, a natural flavone, alleviates inflammatory bowel disease by the inhibition of NLRP3 inflammasome activation via an AhR/Nrf2/NQO1 pathway. Biochem. Pharmacol. 2018, 155, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Kinneer, K.; Bi, Y.; Chan, J.Y.; Kan, Y.W. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): Cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem. J. 2004, 377, 205–213. [Google Scholar] [PubMed]
- Noda, S.; Harada, N.; Hida, A.; Fujii-Kuriyama, Y.; Motohashi, H.; Yamamoto, M. Gene expression of detoxifying enzymes in AhR and Nrf2 compound null mutant mouse. Biochem. Biophys. Res. Commun. 2003, 303, 105–111. [Google Scholar] [CrossRef]
- Furue, M.; Tsuji, G.; Mitoma, C.; Nakahara, T.; Chiba, T.; Morino-Koga, S.; Uchi, H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J. Dermatol. Sci. 2015, 80, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Loertscher, J.A.; Lin, T.M.; Peterson, R.E.; Allen-Hoffmann, B.L. In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin. Toxicol. Sci. 2002, 68, 465–472. [Google Scholar] [CrossRef]
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 2013, 132, 235–249. [Google Scholar] [CrossRef]
- Sutter, C.H.; Bodreddigari, S.; Campion, C.; Wible, R.S.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol. Sci. 2011, 124, 128–137. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997, 34, 605–614. [Google Scholar] [CrossRef]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell. Biol. 2005, 25, 9360–9368. [Google Scholar] [CrossRef]
- Geng, S.; Mezentsev, A.; Kalachikov, S.; Raith, K.; Roop, D.R.; Panteleyev, A.A. Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J. Cell Sci. 2006, 119, 4901–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Tojo, H.; Tomita, S.; Sano, S.; Itami, S.; Hara, M.; Inoue, S.; Horie, K.; Kondoh, G.; Hosokawa, K.; et al. Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function. J. Clin. Investig. 2003, 112, 1372–1382. [Google Scholar] [CrossRef]
- Ju, Q.; Fimmel, S.; Hinz, N.; Stahlmann, R.; Xia, L.; Zouboulis, C.C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters sebaceous gland cell differentiation in vitro. Exp. Dermatol. 2011, 20, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Leu, Y.L.; Yang, S.H.; Chen, H.W.; Wang, C.T.; Pang, J.H. Anti-psoriatic effects of indigo naturalis on the proliferation and differentiation of keratinocytes with indirubin as the active component. J. Dermatol. Sci. 2009, 54, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Rannug, A.; Rannug, U. The tryptophan derivative 6-formylindolo[3,2-b]carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol. 2018, 48, 555–574. [Google Scholar] [CrossRef] [Green Version]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Tanaka, Y.; Ito, T.; Tsuji, G. Implications of tryptophan photoproduct FICZ in oxidative stress and terminal differentiation of keratinocytes. G. Ital. Dermatol. Venereol. 2019, 154, 37–41. [Google Scholar] [CrossRef]
- Kiyomatsu-Oda, M.; Uchi, H.; Morino-Koga, S.; Furue, M. Protective role of 6-formylindolo[3,2-b]carbazole (FICZ), an endogenous ligand for arylhydrocarbon receptor, in chronic mite-induced dermatitis. J. Dermatol. Sci. 2018, 90, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Ito, T.; Chiba, T.; Mitoma, C.; Nakahara, T.; Uchi, H.; Furue, M. The role of the OVOL1-OVOL2 axis in normal and diseased human skin. J. Dermatol. Sci. 2018, 90, 227–231. [Google Scholar] [CrossRef]
- Hong, S.P.; Kim, M.J.; Jung, M.Y.; Jeon, H.; Goo, J.; Ahn, S.K.; Lee, S.H.; Elias, P.M.; Choi, E.H. Biopositive effects of low-dose UVB on epidermis: Coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J. Investig. Dermatol. 2008, 128, 2880–2887. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef]
- Hirano, A.; Goto, M.; Mitsui, T.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidant Artemisia princeps extract enhances the expression of filaggrin and loricrin via the AHR/OVOL1 pathway. Int. J. Mol. Sci. 2017, 18, 1948. [Google Scholar] [CrossRef]
- Hashimoto-Hachiya, A.; Tsuji, G.; Murai, M.; Yan, X.; Furue, M. Upregulation of FLG, LOR, and IVL expression by Rhodiola crenulata root extract via aryl hydrocarbon receptor: Differential involvement of OVOL. Int. J. Mol. Sci. 2018, 19, 1654. [Google Scholar] [CrossRef]
- Van den Bogaard, E.H.; Podolsky, M.A.; Smits, J.P.; Cui, X.; John, C.; Gowda, K.; Desai, D.; Amin, S.G.; Schalkwijk, J.; Perdew, G.H.; et al. Genetic and pharmacological analysis identifies a physiological role for the AHR in epidermal differentiation. J. Investig. Dermatol. 2015, 135, 1320–1328. [Google Scholar] [CrossRef]
- Zhang, W.; Sakai, T.; Matsuda-Hirose, H.; Goto, M.; Yamate, T.; Hatano, Y. Cutaneous permeability barrier function in signal transducer and activator of transcription 6-deficient mice is superior to that in wild-type mice. J. Dermatol. Sci. 2018, 92, 54–61. [Google Scholar] [CrossRef] [Green Version]
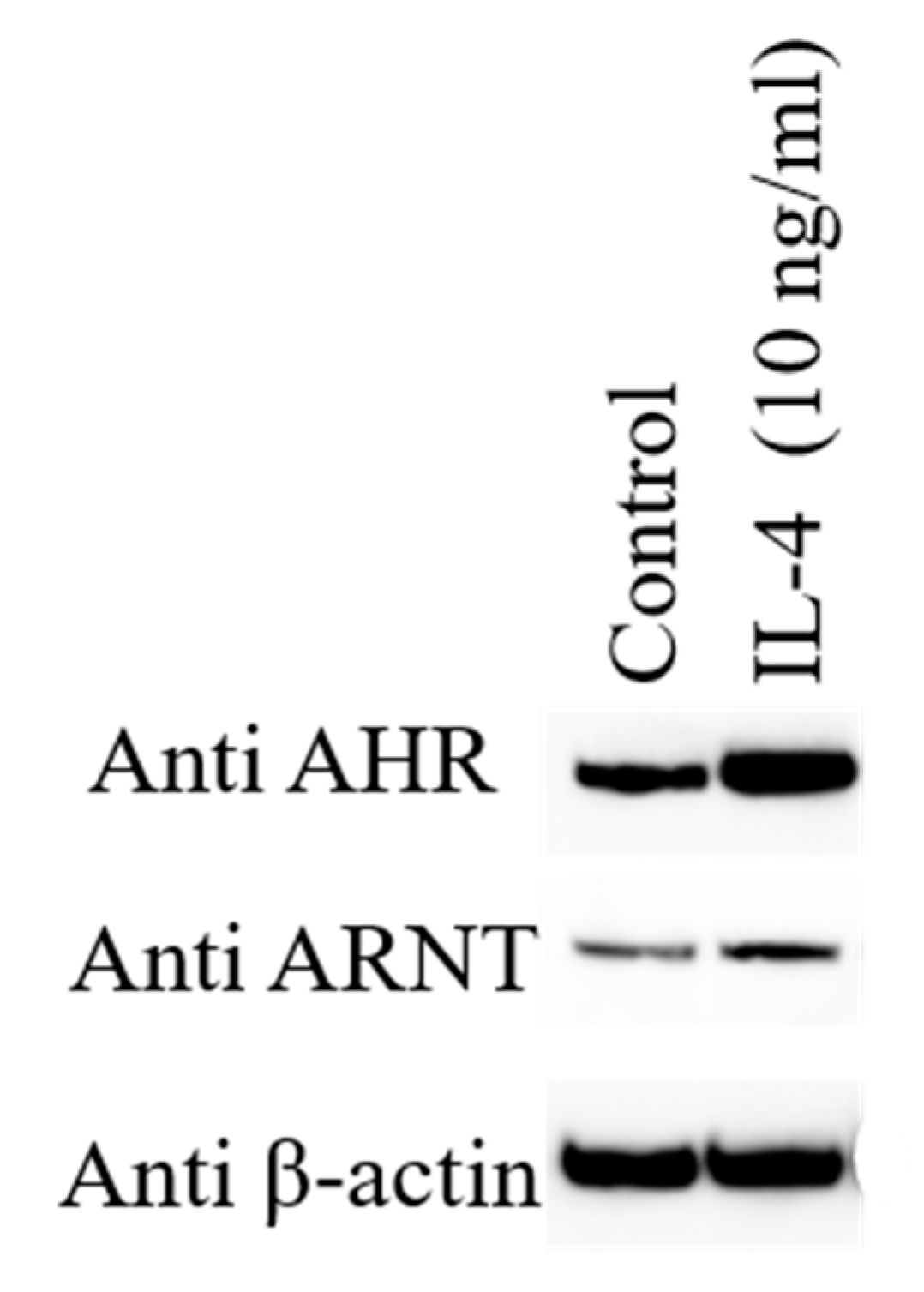
- Tanaka, G.; Kanaji, S.; Hirano, A.; Arima, K.; Shinagawa, A.; Goda, C.; Yasunaga, S.; Ikizawa, K.; Yanagihara, Y.; Kubo, M.; et al. Induction and activation of the aryl hydrocarbon receptor by IL-4 in B cells. Int. Immunol. 2005, 17, 797–805. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Nakahara, T.; Hashimoto-Hachiya, A.; Furue, M.; Tsuji, G. Glyteer, soybean tar, impairs IL-4/Stat6 signaling in murine bone marrow-derived dendritic cells: The basis of its therapeutic effect on atopic dermatitis. Int. J. Mol. Sci. 2018, 19, 1169. [Google Scholar] [CrossRef]
- Schiering, C.; Vonk, A.; Das, S.; Stockinger, B.; Wincent, E. Cytochrome P4501-inhibiting chemicals amplify aryl hydrocarbon receptor activation and IL-22 production in T helper 17 cells. Biochem. Pharmacol. 2018, 151, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Qiu, J.; Bostick, J.W.; Ueda, A.; Schjerven, H.; Li, S.; Jobin, C.; Chen, Z.E.; Zhou, L. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. 2017, 21, 2277–2290. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248. [Google Scholar] [CrossRef]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Holsapple, M.P.; Morris, D.L.; Wood, S.C.; Snyder, N.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: Possible mechanisms. Annu. Rev. Pharmacol. Toxicol. 1991, 31, 73–100. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Funatake, C.J.; Marshall, N.B.; Steppan, L.B.; Mourich, D.V.; Kerkvliet, N.I. Cutting edge: Activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J. Immunol. 2005, 175, 4184–4188. [Google Scholar] [CrossRef]
- Qiu, J.; Guo, X.; Chen, Z.M.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Bisson, W.H.; Kolluri, S.K.; Kerkvliet, N.I. TCDD, FICZ, and other high affinity AhR ligands dose-dependently determine the fate of CD4+ T cell differentiation. Toxicol. Sci. 2018, 161, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Yoon, J.; Jung, S.; Lee, J.; Lee, B.H.; Yu, J. Phenotypes of atopic dermatitis identified by cluster analysis in early childhood. J. Dermatol. 2019, 46, 117–123. [Google Scholar] [CrossRef]
- Arima, K.; Gupta, S.; Gadkari, A.; Hiragun, T.; Kono, T.; Katayama, I.; Demiya, S.; Eckert, L. Burden of atopic dermatitis in Japanese adults: Analysis of data from the 2013 National Health and Wellness Survey. J. Dermatol. 2018, 45, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, A.; Fujita, H.; Arima, K.; Inoue, T.; Dorey, J.; Fukushima, A.; Taguchi, Y. Health-care resource use and current treatment of adult atopic dermatitis patients in Japan: A retrospective claims database analysis. J. Dermatol. 2019, 46, 652–661. [Google Scholar] [CrossRef]
- Jung, H.J.; Bae, J.Y.; Kim, J.E.; Na, C.H.; Park, G.H.; Bae, Y.I.; Shin, M.K.; Lee, Y.B.; Lee, U.H.; Jang, Y.H.; et al. Survey of disease awareness, treatment behavior and treatment satisfaction in patients with atopic dermatitis in Korea: A multicenter study. J. Dermatol. 2018, 45, 1172–1180. [Google Scholar] [CrossRef]
- Komura, Y.; Kogure, T.; Kawahara, K.; Yokozeki, H. Economic assessment of actual prescription of drugs for treatment of atopic dermatitis: Differences between dermatology and pediatrics in large-scale receipt data. J. Dermatol. 2018, 45, 165–174. [Google Scholar] [CrossRef]
- Takeuchi, S.; Oba, J.; Esaki, H.; Furue, M. Non-corticosteroid adherence and itch severity influence perception of itch in atopic dermatitis. J. Dermatol. 2018, 45, 158–164. [Google Scholar] [CrossRef]
- Williams, H.; Stewart, A.; von Mutius, E.; Cookson, W.; Anderson, H.R. Is eczema really on the increase worldwide? J. Allergy Clin. Immunol. 2008, 121, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Iida, K.; Imaji, M.; Nakahara, T. Microbiome analysis of forehead skin in patients with atopic dermatitis and healthy subjects: Implication of Staphylococcus and Corynebacterium. J. Dermatol. 2018, 45, 876–877. [Google Scholar] [CrossRef]
- Iwamoto, K.; Moriwaki, M.; Miyake, R.; Hide, M. Staphylococcus aureus in atopic dermatitis: Strain-specific cell wall proteins and skin immunity. Allergol. Int. 2019, 68, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Kadono, T. “Inflammatory skin march” in atopic dermatitis and psoriasis. Inflamm. Res. 2017, 66, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Z.; Zhong, W.L.; Hu, H.; Chen, X.F.; Zhang, W.; Huang, H.Y.; Yu, B.; Dou, X. Aryl hydrocarbon receptor polymorphisms are associated with dry skin phenotypes in Chinese patients with atopic dermatitis. Clin. Exp. Dermatol. 2019, 44, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Takao, T.; Tsunematsu, R.; Morokuma, S.; Fukushima, K.; Kobayashi, H.; Saito, T.; Furue, M.; Wake, N.; Asanoma, K. Inhibition of AHR transcription by NF1C is affected by a single-nucleotide polymorphism, and is involved in suppression of human uterine endometrial cancer. Oncogene 2013, 32, 4950–4959. [Google Scholar] [CrossRef]
- Liu, G.; Asanoma, K.; Takao, T.; Tsukimori, K.; Uchi, H.; Furue, M.; Kato, K.; Wake, N. Aryl hydrocarbon receptor SNP -130 C/T associates with dioxins susceptibility through regulating its receptor activity and downstream effectors including interleukin. Toxicol. Lett. 2015, 232, 384–392. [Google Scholar] [CrossRef]
- Hong, C.H.; Lee, C.H.; Yu, H.S.; Huang, S.K. Benzopyrene, a major polyaromatic hydrocarbon in smoke fume, mobilizes Langerhans cells and polarizes Th2/17 responses in epicutaneous protein sensitization through the aryl hydrocarbon receptor. Int. Immunopharmacol. 2016, 36, 111–117. [Google Scholar] [CrossRef]
- Kim, H.O.; Kim, J.H.; Chung, B.Y.; Choi, M.G.; Park, C.W. Increased expression of the aryl hydrocarbon receptor in patients with chronic inflammatory skin diseases. Exp. Dermatol. 2014, 23, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Luo, Y.; Zhu, Z.; Zhou, Y.; Sun, L.; Gao, J.; Sun, J.; Wang, G.; Yao, X.; Li, W. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol. 2019, 143, 2108–2119. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Bissonnette, R.; Poulin, Y.; Zhou, Y.; Tan, J.; Hong, H.C.; Webster, J.; Ip, W.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to severe atopic dermatitis: Results from a 12-week, multicentre, randomized, placebo-controlled double-blind trial. Br. J. Dermatol. 2012, 166, 853–860. [Google Scholar] [CrossRef]
- Richardson, W.H.; Schmidt, T.M.; Nealson, K.H. Identification of an anthraquinone pigment and a hydroxystilbene antibiotic from Xenorhabdus luminescens. Appl. Environ. Microbiol. 1988, 54, 1602–16055. [Google Scholar]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef]
- Zang, Y.N.; Jiang, D.L.; Cai, L.; Chen, X.; Wang, Q.; Xie, Z.W.; Liu, Y.; Zhang, C.Y.; Jing, S.; Chen, G.H.; et al. Use of a dose-response model to guide future clinical trial of Benvitimod cream to treat mild and moderate psoriasis. Int. J. Clin. Pharmacol. Ther. 2016, 54, 87–95. [Google Scholar] [CrossRef]
- Bissonnette, R.; Vasist, L.S.; Bullman, J.N.; Collingwood, T.; Chen, G.; Maeda-Chubachi, T. Systemic pharmacokinetics, safety, and preliminary efficacy of topical AhR agonist Tapinarof: Results of a phase 1 study. Clin. Pharmacol. Drug Dev. 2018, 7, 524–531. [Google Scholar] [CrossRef]
- Miake, S.; Tsuji, G.; Takemura, M.; Hashimoto-Hachiya, A.; Vu, Y.H.; Furue, M.; Nakahara, T. IL-4 augments IL-31/IL-31 receptor alpha interaction leading to enhanced Ccl17 and Ccl22 production in dendritic cells: Implications for atopic dermatitis. Int. J. Mol. Sci. 2019, 20, 4053. [Google Scholar] [CrossRef]
- Koch, S.; Stroisch, T.J.; Vorac, J.; Herrmann, N.; Leib, N.; Schnautz, S.; Kirins, H.; Forster, I.; Weighardt, H.; Bieber, T. AhR mediates an anti-inflammatory feedback mechanism in human Langerhans cells involving FcεRI and IDO. Allergy 2017, 72, 1686–1693. [Google Scholar] [CrossRef]
- Edamitsu, T.; Taguchi, K.; Kobayashi, E.H.; Okuyama, R.; Yamamoto, M. Aryl hydrocarbon receptor directly regulates artemin gene expression. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef]
- Morita, A. Current developments in phototherapy for psoriasis. J. Dermatol. 2018, 45, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Salvador, J.M.; Pérez-Ferriols, A. Phototherapy in Atopic Dermatitis. Adv. Exp. Med. Biol. 2017, 996, 279–286. [Google Scholar]
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Furue, M.; Kadono, T. The contribution of IL-17 to the development of autoimmunity in psoriasis. Innate Immun. 2019, 25, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.; Okuyama, R.; Seki, T.; Kobayashi, A.; Oiso, N.; Muto, M.; Nakagawa, H.; Kawada, A. Epidemiological survey of patients with psoriasis in Matsumoto city, Nagano Prefecture, Japan. J. Dermatol. 2018, 45, 314–317. [Google Scholar] [CrossRef]
- Ito, T.; Takahashi, H.; Kawada, A.; Iizuka, H.; Nakagawa, H. Epidemiological survey from 2009 to 2012 of psoriatic patients in Japanese Society for Psoriasis Research. J. Dermatol. 2018, 45, 293–301. [Google Scholar] [CrossRef]
- Ichiyama, S.; Ito, M.; Funasaka, Y.; Abe, M.; Nishida, E.; Muramatsu, S.; Nishihara, H.; Kato, H.; Morita, A.; Imafuku, S.; et al. Assessment of medication adherence and treatment satisfaction in Japanese patients with psoriasis of various severities. J. Dermatol. 2018, 45, 727–731. [Google Scholar] [CrossRef]
- Takahashi, H.; Satoh, K.; Takagi, A.; Iizuka, H. Cost-efficacy and pharmacoeconomics of psoriatic patients in Japan: Analysis from a single outpatient clinic. J. Dermatol. 2019, 46, 478–481. [Google Scholar] [CrossRef]
- Komatsu-Fujii, T.; Honda, T.; Otsuka, A.; Kabashima, K. Improvement of nail lesions in a patient with psoriatic arthritis by switching the treatment from an anti-interleukin-17A antibody to an anti-tumor necrosis factor-α antibody. J. Dermatol. 2019, 46, e158–e160. [Google Scholar] [CrossRef]
- Tsuruta, N.; Narisawa, Y.; Imafuku, S.; Ito, K.; Yamaguchi, K.; Miyagi, T.; Takahashi, K.; Fukamatsu, H.; Morizane, S.; Koketsu, H.; et al. Cross-sectional multicenter observational study of psoriatic arthritis in Japanese patients: Relationship between skin and joint symptoms and results of treatment with tumor necrosis factor-α inhibitors. J. Dermatol. 2019, 46, 193–198. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ohtsuki, M.; Sano, S.; Morita, A.; Igarashi, A.; Okuyama, R.; Kawada, A. Late-onset psoriatic arthritis in Japanese patients. J. Dermatol. 2019, 46, 169–170. [Google Scholar] [CrossRef]
- Chujo, S.; Asahina, A.; Itoh, Y.; Kobayashi, K.; Sueki, H.; Ishiji, T.; Umezawa, Y.; Nakagawa, H. New onset of psoriasis during nivolumab treatment for lung cancer. J. Dermatol. 2018, 45, e55–e56. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Nakahara, T.; Furue, M. Autoimmunity and autoimmune co-morbidities in psoriasis. Immunology 2018, 154, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.H.; Hu, H.Y.; Chang, Y.T.; Li, C.P.; Wu, C.Y. Psoriasis is associated with increased risk of bullous pemphigoid: A nationwide population-based cohort study in Taiwan. J. Dermatol. 2019, 46, 604–609. [Google Scholar] [CrossRef]
- Ichiyama, S.; Hoashi, T.; Kanda, N.; Hashimoto, H.; Matsushita, M.; Nozawa, K.; Ueno, T.; Saeki, H. Psoriasis vulgaris associated with systemic lupus erythematosus successfully treated with apremilast. J. Dermatol. 2019, 46, e219–e221. [Google Scholar] [CrossRef]
- Kamata, M.; Asano, Y.; Shida, R.; Maeda, N.; Yoshizaki, A.; Miyagaki, T.; Kawashima, T.; Tada, Y.; Sato, S. Secukinumab decreased circulating anti-BP180-NC16a autoantibodies in a patient with coexisting psoriasis vulgaris and bullous pemphigoid. J. Dermatol. 2019, 46, e216–e217. [Google Scholar] [CrossRef]
- Masaki, S.; Bayaraa, B.; Imafuku, S. Prevalence of inflammatory bowel disease in Japanese psoriatic patients. J. Dermatol. 2019, 46, 590–594. [Google Scholar] [CrossRef]
- Chiu, H.Y.; Chang, W.L.; Shiu, M.N.; Huang, W.F.; Tsai, T.F. Psoriasis is associated with a greater risk for cardiovascular procedure and surgery in patients with hypertension: A nationwide cohort study. J. Dermatol. 2018, 45, 1381–1388. [Google Scholar] [CrossRef]
- Momose, M.; Asahina, A.; Fukuda, T.; Sakuma, T.; Umezawa, Y.; Nakagawa, H. Evaluation of epicardial adipose tissue volume and coronary artery calcification in Japanese patients with psoriasis vulgaris. J. Dermatol. 2018, 45, 1349–1352. [Google Scholar] [CrossRef]
- Takamura, S.; Takahashi, A.; Inoue, Y.; Teraki, Y. Effects of tumor necrosis factor-α, interleukin-23 and interleukin-17A inhibitors on bodyweight and body mass index in patients with psoriasis. J. Dermatol. 2018, 45, 1130–1134. [Google Scholar] [CrossRef]
- Han, J.H.; Lee, J.H.; Han, K.D.; Kim, H.N.; Bang, C.H.; Park, Y.M.; Lee, J.Y.; Kim, T.Y. Increased risk of psoriasis in subjects with abdominal obesity: A nationwide population-based study. J. Dermatol. 2019, 46, 695–701. [Google Scholar] [CrossRef]
- Yamazaki, F.; Takehana, K.; Tamashima, M.; Okamoto, H. Improvement in abnormal coronary arteries estimated by coronary computed tomography angiography after secukinumab treatment in a Japanese psoriatic patient. J. Dermatol. 2019, 46, e51–e52. [Google Scholar] [CrossRef]
- Bayaraa, B.; Imafuku, S. Sustainability and switching of biologics for psoriasis and psoriatic arthritis at Fukuoka University Psoriasis Registry. J. Dermatol. 2019, 46, 389–398. [Google Scholar] [CrossRef]
- Nakajima, K.; Sano, S. Mouse models of psoriasis and their relevance. J. Dermatol. 2018, 45, 252–263. [Google Scholar] [CrossRef]
- Ogawa, E.; Sato, Y.; Minagawa, A.; Okuyama, R. Pathogenesis of psoriasis and development of treatment. J. Dermatol. 2018, 45, 264–272. [Google Scholar] [CrossRef]
- Kamata, M.; Tada, Y. Safety of biologics in psoriasis. J. Dermatol. 2018, 45, 279–286. [Google Scholar] [CrossRef]
- Tada, Y.; Ishii, K.; Kimura, J.; Hanada, K.; Kawaguchi, I. Patient preference for biologic treatments of psoriasis in Japan. J. Dermatol. 2019, 46, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Bayaraa, B.; Imafuku, S. Relationship between environmental factors, age of onset and familial history in Japanese patients with psoriasis. J. Dermatol. 2018, 45, 715–718. [Google Scholar] [CrossRef]
- Elder, J.T. Expanded genome-wide association study meta-analysis of psoriasis expands the catalog of common psoriasis-associated variants. J. Investig. Dermatol. Symp. Proc. 2018, 19, S77–S78. [Google Scholar] [CrossRef]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef]
- Smith, S.H.; Peredo, C.E.; Takeda, Y.; Bui, T.; Neil, J.; Rickard, D.; Millerman, E.; Therrien, J.P.; Nicodeme, E.; Brusq, J.M.; et al. Development of a topical treatment for psoriasis targeting RORγ: From bench to skin. PLoS ONE 2016, 11, e0147979. [Google Scholar] [CrossRef]
- Cochez, P.M.; Michiels, C.; Hendrickx, E.; Van Belle, A.B.; Lemaire, M.M.; Dauguet, N.; Warnier, G.; de Heusch, M.; Togbe, D.; Ryffel, B.; et al. AhR modulates the IL-22-producing cell proliferation/recruitment in imiquimod-induced psoriasis mouse model. Eur. J. Immunol. 2016, 46, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Mescher, M.; Tigges, J.; Rolfes, K.M.; Shen, A.L.; Yee, J.S.; Vogeley, C.; Krutmann, J.; Bradfield, C.A.; Lang, D.; Haarmann-Stemmann, T. The Toll-like receptor agonist imiquimod is metabolized by aryl hydrocarbon receptor-regulated cytochrome P450 enzymes in human keratinocytes and mouse liver. Arch. Toxicol. 2019, 93, 1917–1926. [Google Scholar] [CrossRef]
- Beranek, M.; Fiala, Z.; Kremlacek, J.; Andrys, C.; Krejsek, J.; Hamakova, K.; Palicka, V.; Borska, L. Serum levels of aryl hydrocarbon receptor, cytochromes P450 1A1 and 1B1 in patients with exacerbated psoriasis vulgaris. Folia Biol. (Praha) 2018, 64, 97–102. [Google Scholar] [PubMed]
- Bissonnette, R.; Bolduc, C.; Maari, C.; Nigen, S.; Webster, J.M.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to moderate psoriasis: Results from a randomized double-blind placebo-controlled, phase II trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1516–1521. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. https://doi.org/10.3390/ijms20215424
Furue M, Hashimoto-Hachiya A, Tsuji G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. International Journal of Molecular Sciences. 2019; 20(21):5424. https://doi.org/10.3390/ijms20215424
Chicago/Turabian StyleFurue, Masutaka, Akiko Hashimoto-Hachiya, and Gaku Tsuji. 2019. "Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis" International Journal of Molecular Sciences 20, no. 21: 5424. https://doi.org/10.3390/ijms20215424
APA StyleFurue, M., Hashimoto-Hachiya, A., & Tsuji, G. (2019). Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. International Journal of Molecular Sciences, 20(21), 5424. https://doi.org/10.3390/ijms20215424