RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
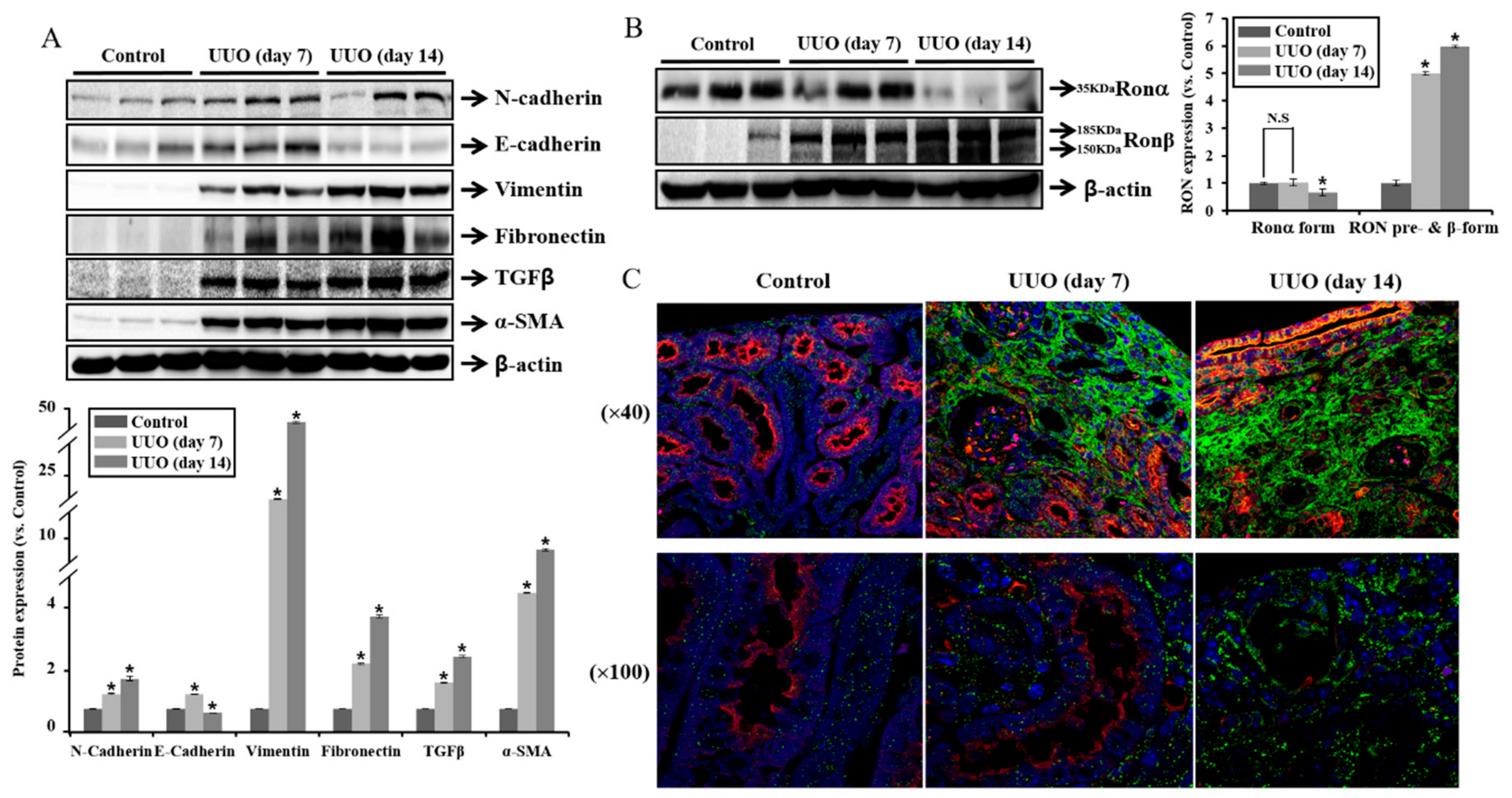
2.1. Expression of RON Is Associated with Fibrosis in UUO Mouse Model
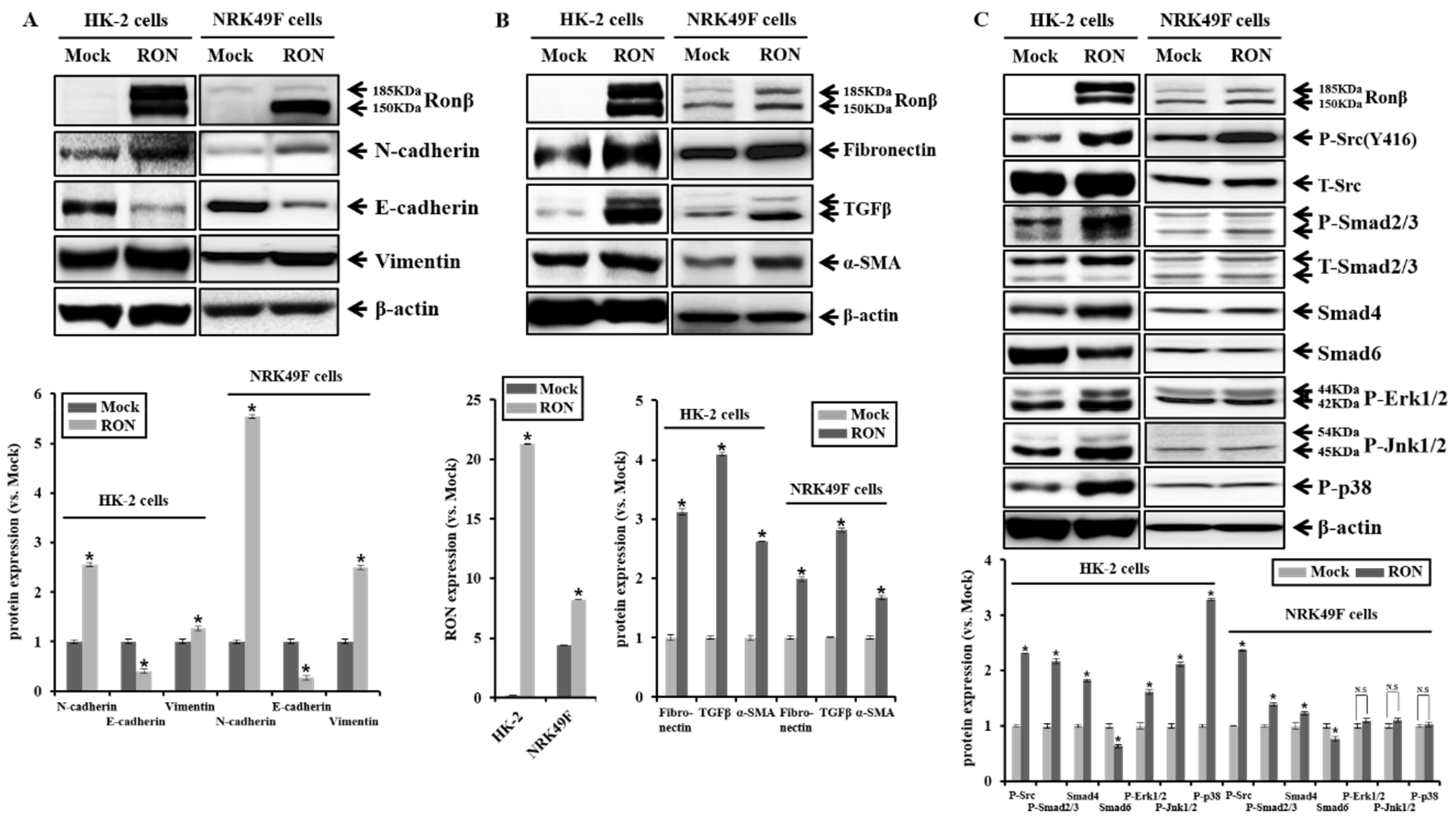
2.2. Effect of RON Overexpression in Proximal Tubular HK-2 and Interstitial Fibroblasts NRK49F Cells
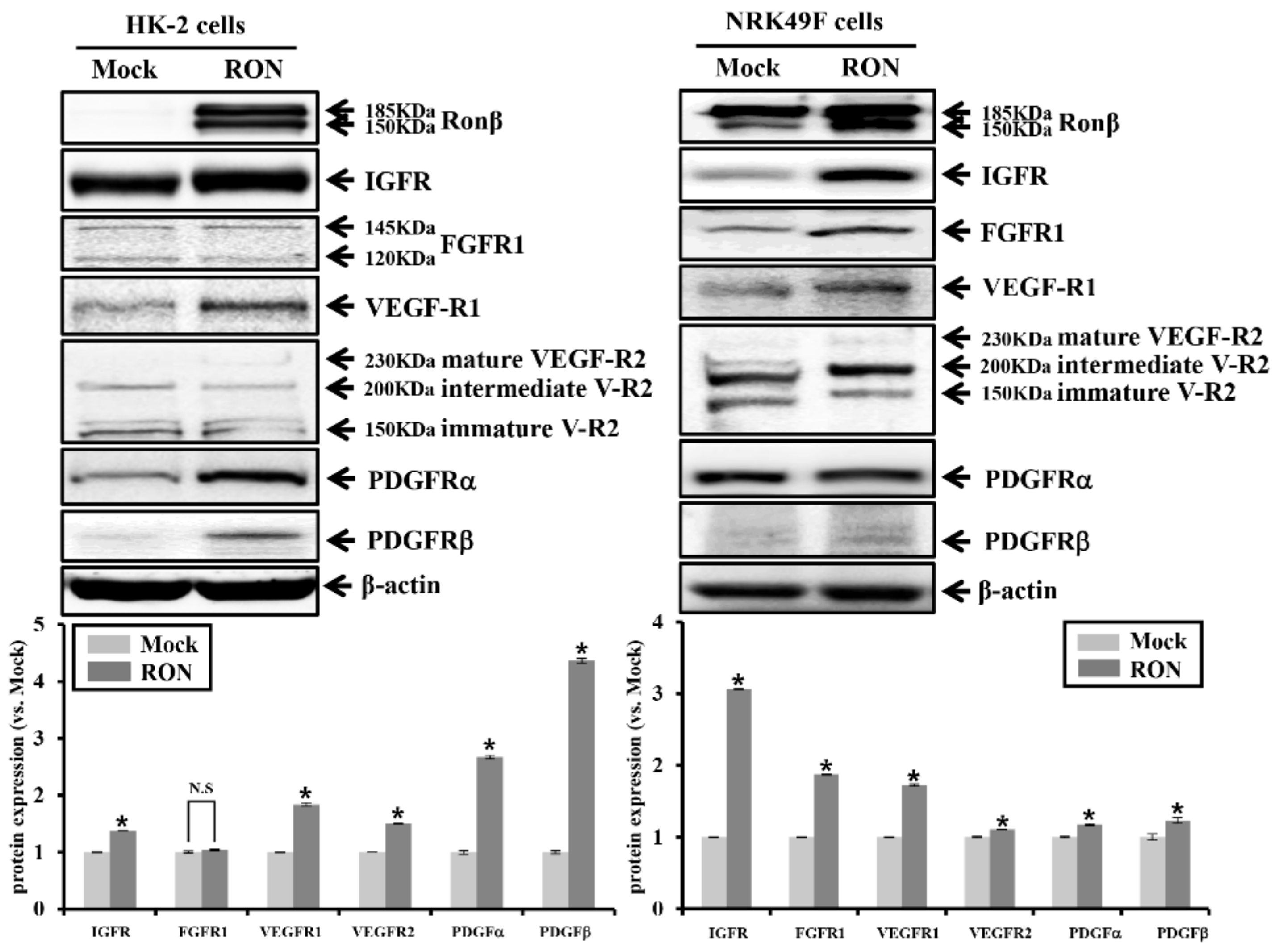
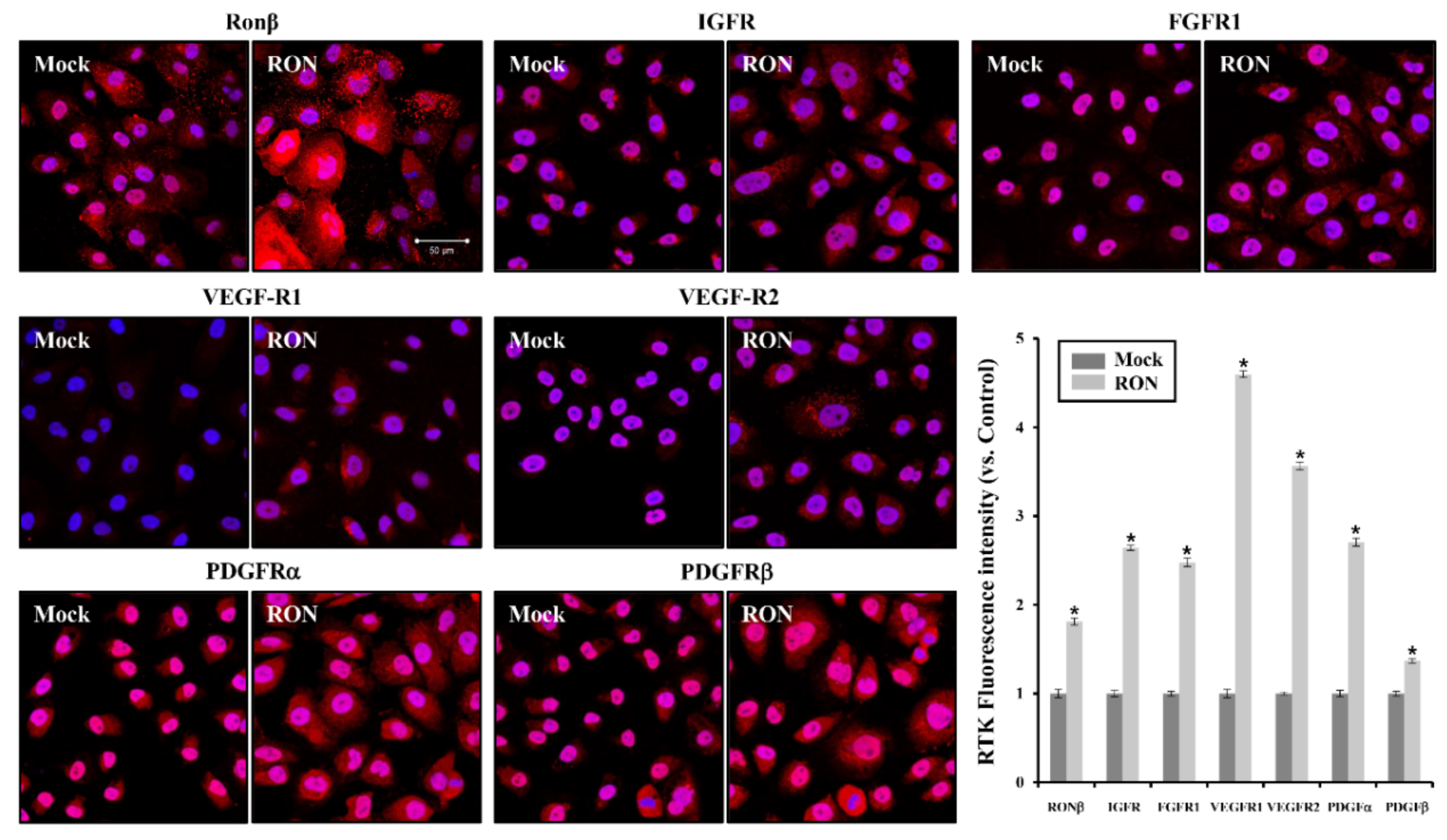
2.3. Effects of RON on Other RTKs in Proximal Tubular HK-2 and Interstitial Fibroblast NRK49F Cells
2.4. Effect of RON siRNA on EMT, Pro-Fibrotic Marker, Src Signaling Pathway in HK-2 and NRK49F Cells
2.5. Effects of Src siRNA on EMT, Pro-Fibrotic Marker, and RTKs in HK-2 and NRK49F Cells
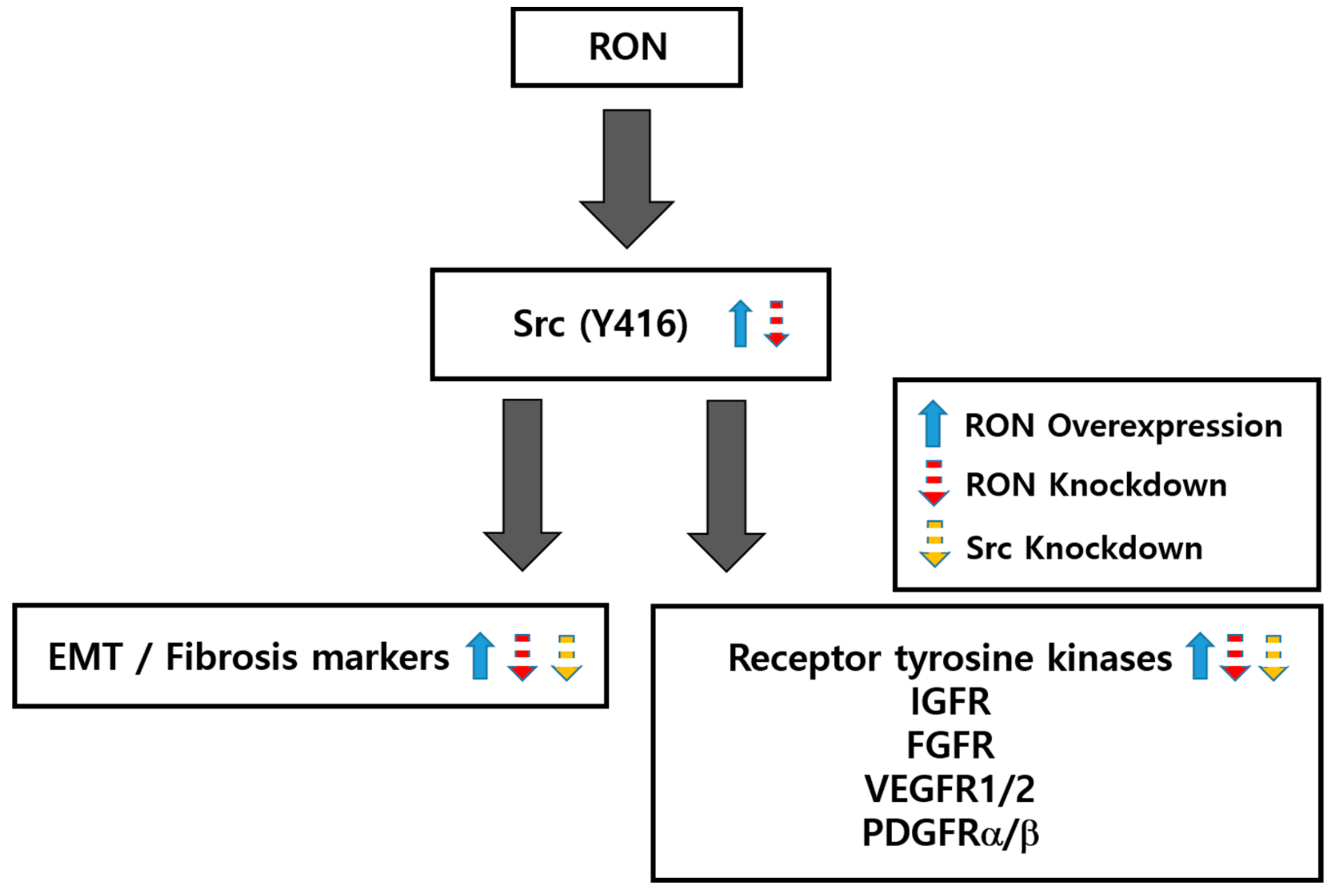
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Animals
4.3. Cell Culture
4.4. Immunohistofluorescence (IHF)
4.5. Stable Cell Lines
4.6. Transient Transfection of Plasmid Construct, RON
4.7. siRNA Knockdown
4.8. Western Blot Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Prabhakaran, D.; Anand, S.; Watkins, D.; Gaziano, T.; Wu, Y.; Mbanya, J.C.; Nugent, R.; Ajay, V.S.; Afshin, A.; Adler, A.; et al. Cardiovascular, respiratory, and related disorders: Key messages from Disease Control Priorities, 3rd edition. Lancet 2018, 391, 1224–1236. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J.; Warnock, D.G.; Delanaye, P. The global burden of chronic kidney disease: Estimates, variability and pitfalls. Nat. Rev. Nephrol. 2017, 13, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef]
- Chen, S.C.; Huang, J.C.; Su, H.M.; Chiu, Y.W.; Chang, J.M.; Hwang, S.J.; Chen, H.C. Prognostic Cardiovascular Markers in Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 1388–1407. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Leaf, I.A.; Duffield, J.S. What can target kidney fibrosis? Nephrol. Dial. Transplant. 2017, 32, i89–i97. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Zeisberg, M.; Maeshima, Y.; Mosterman, B.; Kalluri, R. Renal fibrosis. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am. J. Pathol. 2002, 160, 2001–2008. [Google Scholar] [CrossRef]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhuang, S. Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis. Int. J. Mol. Sci. 2016, 17, 972. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Khaibullina, A.; Adjei, E.A.; Afangbedji, N.; Ivanov, A.; Kumari, N.; Almeida, L.E.F.; Quezado, Z.M.N.; Nekhai, S.; Jerebtsova, M. RON kinase inhibition reduces renal endothelial injury in sickle cell disease mice. Haematologica 2018, 103, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Floege, J.; Eitner, F.; Alpers, C.E. A new look at platelet-derived growth factor in renal disease. J. Am. Soc. Nephrol. 2008, 19, 12–23. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; Van Kooten, C.; Muller, G. TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 2001, 59, 579–592. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Wang, M.H.; Wang, D.; Chen, Y.Q. Oncogenic and invasive potentials of human macrophage-stimulating protein receptor, the RON receptor tyrosine kinase. Carcinogenesis 2003, 24, 1291–1300. [Google Scholar] [CrossRef]
- Camp, E.R.; Liu, W.; Fan, F.; Yang, A.; Somcio, R.; Ellis, L.M. RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann. Surg. Oncol. 2005, 12, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.R.; Yang, A.; Gray, M.J.; Fan, F.; Hamilton, S.R.; Evans, D.B.; Hooper, A.T.; Pereira, D.S.; Hicklin, D.J.; Ellis, L.M. Tyrosine kinase receptor RON in human pancreatic cancer: Expression, function, and validation as a target. Cancer 2007, 109, 1030–1039. [Google Scholar] [CrossRef]
- Liu, F.; Wang, L.; Qi, H.; Wang, J.; Wang, Y.; Jiang, W.; Xu, L.; Liu, N.; Zhuang, S. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin. Sci. 2017, 131, 2125–2143. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Sweeney, W.E., Jr.; Wang, X.; Qian, Q.; Harris, P.C.; Frost, P.; Avner, E.D. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han: SPRD rats. Kidney Int. 2003, 64, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Chade, A.R. Vascular Endothelial Growth Factor Therapy for the Kidney: Are We There Yet? J. Am. Soc. Nephrol. 2016, 27, 1–3. [Google Scholar] [CrossRef]
- Benight, N.M.; Waltz, S.E. Ron receptor tyrosine kinase signaling as a therapeutic target. Expert Opin. Ther. Targets 2012, 16, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef]
- Arman, E.; Haffner-Krausz, R.; Chen, Y.; Heath, J.K.; Lonai, P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl. Acad. Sci. USA 1998, 95, 5082–5087. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.X.; Wynshaw-Boris, A.; Shen, M.M.; Daugherty, C.; Ornitz, D.M.; Leder, P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994, 8, 3045–3057. [Google Scholar] [CrossRef]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.P.; Guadagnini, D.; Goncalves, F.L.; Barini, R.; Saad, M.J.; Schmidt, A.F.; Sbragia, L. Assessment of the expression of IRbeta, IRS-1, IRS-2 and IGF-IRbeta in a rat model of intrauterine growth restriction. Fetal Diagn. Ther. 2010, 28, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.; Chan, T.M. Pathophysiological changes to the peritoneal membrane during PD-related peritonitis: The role of mesothelial cells. Mediat. Inflamm. 2012, 2012, 484167. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cabrera, M. Mesenchymal Conversion of Mesothelial Cells Is a Key Event in the Pathophysiology of the Peritoneum during Peritoneal Dialysis. Adv. Med. 2014, 2014, 473134. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251. [Google Scholar] [CrossRef] [PubMed]
- Danilkovitch-Miagkova, A. Oncogenic signaling pathways activated by RON receptor tyrosine kinase. Curr. Cancer Drug Targets 2003, 3, 31–40. [Google Scholar] [CrossRef]
- Ronsin, C.; Muscatelli, F.; Mattei, M.G.; Breathnach, R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene 1993, 8, 1195–1202. [Google Scholar]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. C-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.; Thomas, M.L. Recent advances in lymphocyte signaling and regulation. Front. Biosci. 1997, 2, d207–d221. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ma, L.; Zhou, X.; Ponnusamy, M.; Tang, J.; Zhuang, M.A.; Tolbert, E.; Bayliss, G.; Bai, J.; Zhuang, S. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 2016, 89, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromann, P.A.; Korkaya, H.; Courtneidge, S.A. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhuang, S. Src family kinases in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F721–F728. [Google Scholar] [CrossRef] [Green Version]
- Zacchia, M.; Tian, X.; Zona, E.; Alpern, R.J.; Preisig, P.A. Acid Stimulation of the Citrate Transporter NaDC-1 Requires Pyk2 and ERK1/2 Signaling Pathways. J. Am. Soc. Nephrol. 2018, 29, 1720–1730. [Google Scholar] [CrossRef]
- Kopp, J.B.; Heymann, J. C-Src is in the effector pathway linking uPAR and podocyte injury. J. Clin. Investig. 2019, 129, 1827–1829. [Google Scholar] [CrossRef]
- Xiangming, X.; Yun, Q.; Guoliang, Z.; Jianjiang, L.; Lisong, T. Mechanisms of RON-mediated epithelial-mesenchymal transition in MDCK cells through the MAPK pathway. Braz. J. Med. Biol. Res. 2011, 44, 634–641. [Google Scholar]
- Zhang, D.Y.; Krell, P.J.; Feng, Q.L. Two lepidopteran cell lines stably transformed by the abc transporter gene pdr5 show tolerance to diacetoxyscirpenol. In Vitro Cell Dev. Biol. Anim. 2006, 42, 27–32. [Google Scholar] [CrossRef]
- Kim, C.S.; Joo, S.Y.; Lee, K.E.; Choi, J.S.; Bae, E.H.; Ma, S.K.; Kim, S.H.; Lee, J.; Kim, S.W. Paricalcitol attenuates 4-hydroxy-2-hexenal-induced inflammation and epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. PLoS ONE 2013, 8, e63186. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.S.; Choi, H.-I.; Kim, D.-H.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells. Int. J. Mol. Sci. 2019, 20, 5489. https://doi.org/10.3390/ijms20215489
Park JS, Choi H-I, Kim D-H, Kim CS, Bae EH, Ma SK, Kim SW. RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells. International Journal of Molecular Sciences. 2019; 20(21):5489. https://doi.org/10.3390/ijms20215489
Chicago/Turabian StylePark, Jung Sun, Hoon-In Choi, Dong-Hyun Kim, Chang Seong Kim, Eun Hui Bae, Seong Kwon Ma, and Soo Wan Kim. 2019. "RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells" International Journal of Molecular Sciences 20, no. 21: 5489. https://doi.org/10.3390/ijms20215489
APA StylePark, J. S., Choi, H. -I., Kim, D. -H., Kim, C. S., Bae, E. H., Ma, S. K., & Kim, S. W. (2019). RON Receptor Tyrosine Kinase Regulates Epithelial Mesenchymal Transition and the Expression of Pro-Fibrotic Markers via Src/Smad Signaling in HK-2 and NRK49F Cells. International Journal of Molecular Sciences, 20(21), 5489. https://doi.org/10.3390/ijms20215489