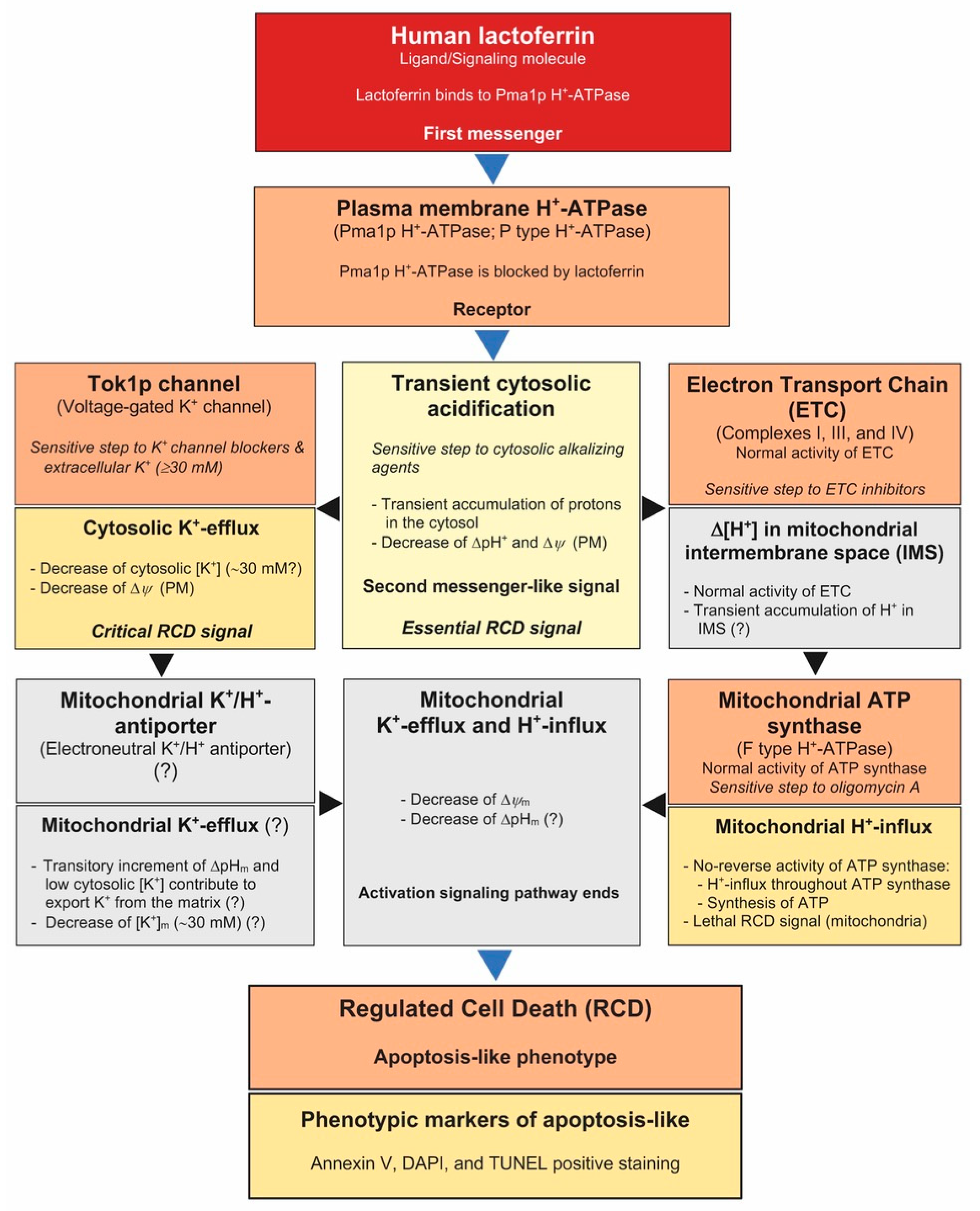
Cytosolic Acidification Is the First Transduction Signal of Lactoferrin-Induced Regulated Cell Death Pathway

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
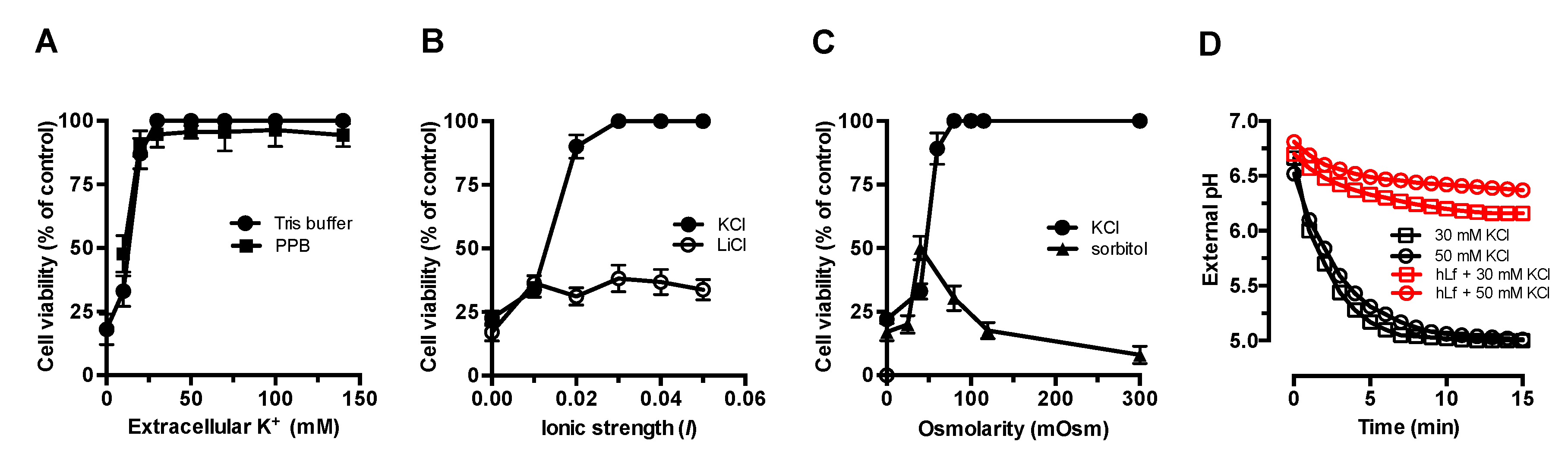
2.1. Effect of Extracellular Factors on the RCD Induced by Lactoferrin
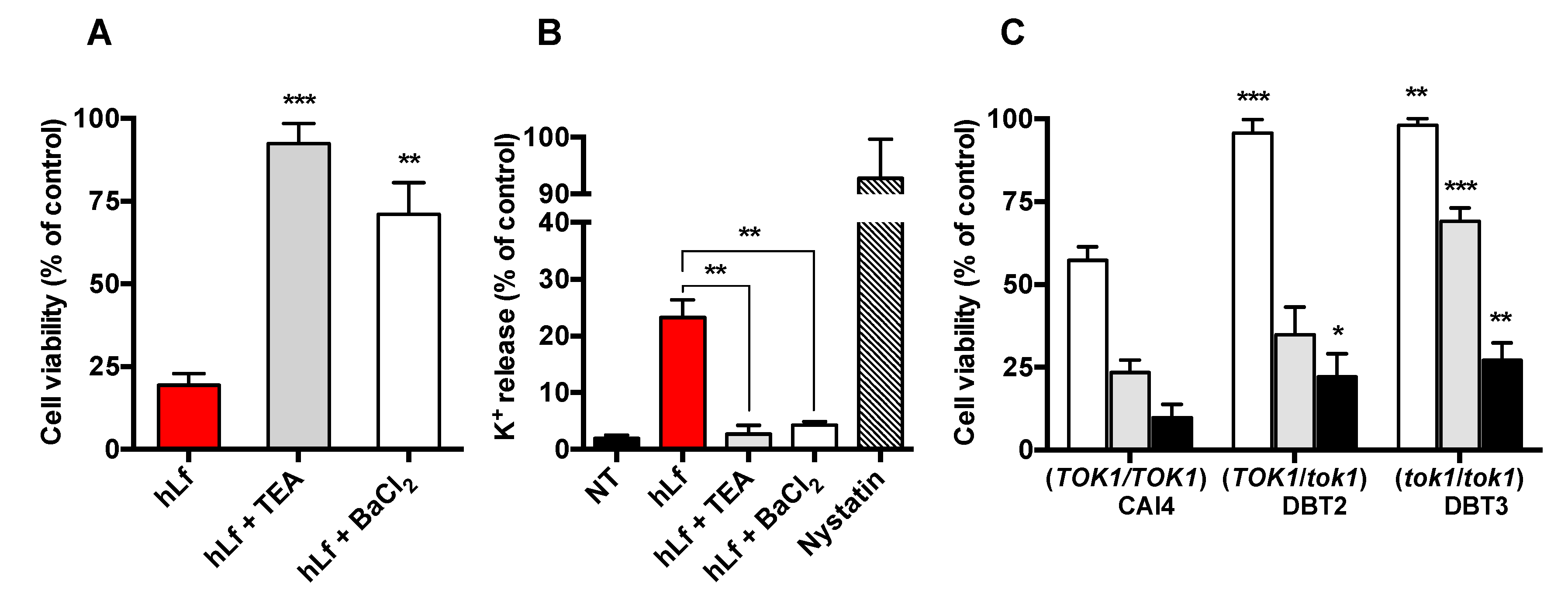
2.2. Identification of K+ Transporters in the RCD Process
2.3. Effect of Cellular K+ Depletion on RCD Triggering
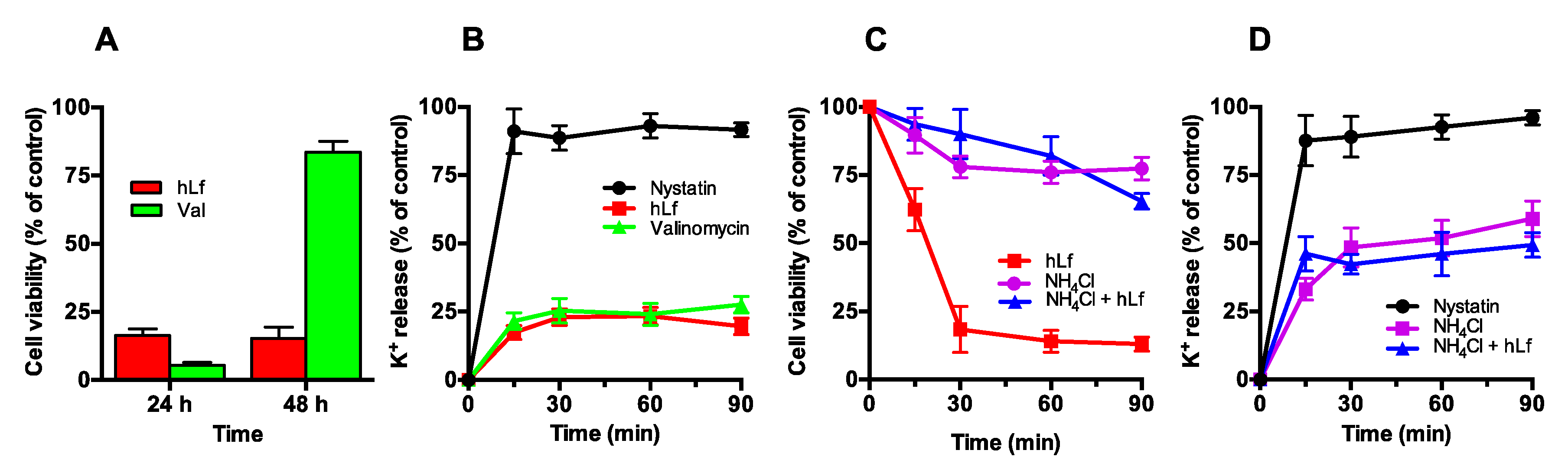
2.3.1. Effect of Valinomycin-Mediated K+ Release on Cell Viability
2.3.2. Effect of NH4Cl-Mediated K+ Extrusion on Cell Viability
2.3.3. Effect of Nigericin-Mediated K+/H+ Exchange on Cell Viability
2.3.4. Effect of Piericidin A on the Lactoferrin Induced K+-efflux
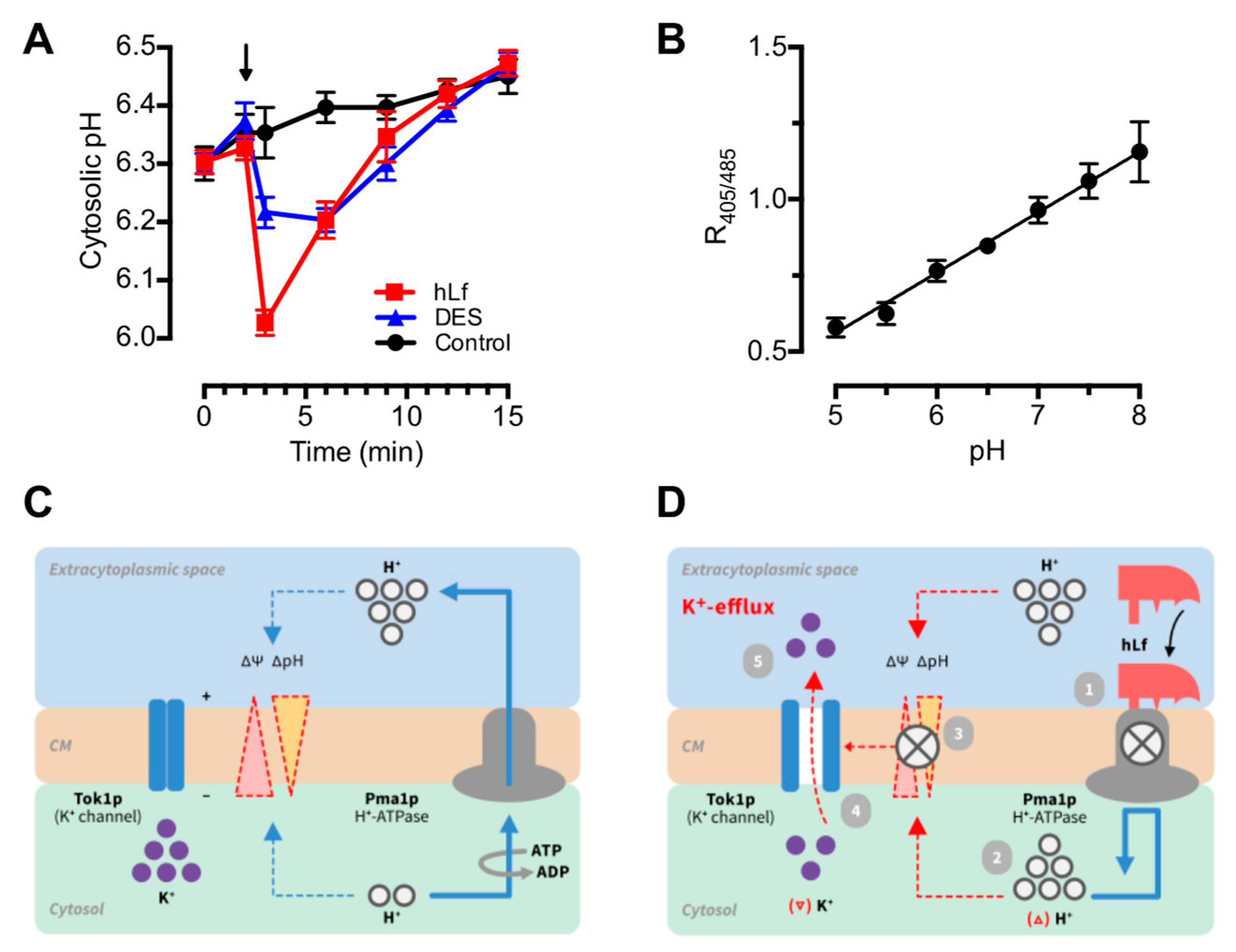
2.4. Evaluation of the Role of Cytosolic pH in Regulated Cell Death Triggering
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Strains and Growth Conditions
4.3. Antifungal Assays
4.4. Measurement of Extracellular Potassium
4.5. Glucose-dependent External Acidification
4.6. Measurement of Cytosolic pH
4.7. Other Methods
4.8. Statistical Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klotz, S.A.; Chasin, B.S.; Powell, B.; Gaur, N.K.; Lipke, P.N. Polymicrobial bloodstream infections involving Candida species: Analysis of patients and review of the literature. Diagn. Microbiol. Infect. Dis. 2007, 59, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Andrés, M.T.; Viejo-Diaz, M.; Fierro, J.F. Human lactoferrin induces apoptosis-like cell death in Candida albicans: Critical role of K+-channel-mediated K+ efflux. Antimicrob. Agents Chemother. 2008, 52, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.E.; Carter, D.A. The antifungal activity of lactoferrin and its derived peptides: Mechanisms of action and synergy with drugs against fungal pathogens. Front. Microbiol. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Andrés, M.T.; Acosta-Zaldívar, M.; Fierro, J.F. Antifungal mechanism of action of lactoferrin: Identification of H+-ATPase (P3A-type) as a new apoptotic-cell membrane receptor. Antimicrob. Agents Chemother. 2016, 60, 4206–4216. [Google Scholar] [CrossRef]
- Peña, A. Studies on the mechanism of K+ transport in yeast. Arch. Biochem. Biophys. 1975, 167, 397–409. [Google Scholar] [CrossRef]
- Serrano, R. Structure and function of plasma membrane ATPase. Ann. Rev. Plant. Physiol. Plant Mol. Biol. 1989, 40, 61–94. [Google Scholar] [CrossRef]
- Ariño, J.; Ramos, J.; Sychrová, H. Monovalent cation transporters at the plasma membrane in yeasts. Yeast 2019, 36, 177–193. [Google Scholar] [CrossRef]
- Acosta-Zaldívar, M.; Andrés, M.T.; Rego, A.; Pereira, C.S.; Fierro, J.F.; Côrte-Real, M. Human lactoferrin triggers a mitochondrial- and caspase-dependent regulated cell death in Saccharomyces cerevisiae. Apoptosis 2016, 21, 163–173. [Google Scholar] [CrossRef]
- Bortner, C.D.; Hughes, F.M., Jr.; Cidlowski, J.A. A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 1997, 272, 32436–32442. [Google Scholar] [CrossRef]
- Yu, S.P.; Yeh, C.H.; Sensi, S.L.; Gwag, B.J.; Canzoniero, L.M.; Farhangrazi, Z.S.; Ying, H.S.; Tian, M.; Dugan, L.L.; Choi, D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997, 278, 114–117. [Google Scholar] [CrossRef]
- Dallaporta, B.; Hirsch, T.; Susin, S.A.; Zamzami, N.; Larochette, N.; Brenner, C.; Marzo, I.; Kroemer, G. Potassium leakage during the apoptotic degradation phase. J. Immunol. 1998, 160, 5605–5615. [Google Scholar] [PubMed]
- Hughes, F.M.; Cidlowski, J.A. Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv. Enzyme Regul. 1999, 39, 157–171. [Google Scholar] [CrossRef]
- Park, I.S.; Kim, J.E. Potassium efflux during apoptosis. J. Biochem. Mol. Biol. 2002, 35, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 2003, 70, 363–386. [Google Scholar] [CrossRef]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. K+ channels in apoptosis. J. Membr. Biol. 2006, 209, 3–20. [Google Scholar] [CrossRef]
- Peters, J.; Chin, C.-K. Potassium loss is involved in tobacco cell death induced by palmitoleic acid and ceramide. Archiv. Biochem. Biophys. 2007, 465, 180–186. [Google Scholar] [CrossRef]
- Hoeberichts, F.A.; Pérez-Valle, J.; Montesinos, C.; Mulet, J.M.; Planes, M.D.; Hueso, G.; Yenush, L.; Sharma, S.C.; Serrano, R. The role of K+ and H+ transport systems during glucose- and H2O2-induced cell death in Saccharomyces cerevisiae. Yeast 2010, 27, 713–725. [Google Scholar] [CrossRef]
- Klein, B.; Wörndl, K.; Lütz-Meindl, U.; Kerschbaum, H.H. Perturbation of intracellular K+ homeostasis with valinomycin promotes cell death by mitochondrial swelling and autophagic processes. Apoptosis 2011, 16, 1101–1117. [Google Scholar] [CrossRef]
- Demidchik, V.; Straltsova, D.; Medvedev, S.S.; Pozhvanov, G.A.; Sokolik, A.; Yurin, V. Stress-induced electrolyte leakage: The role of K+-permeable channels and involvement in programmed cell death and metabolic adjustment. J. Exp. Bot. 2014, 65, 1259–1270. [Google Scholar] [CrossRef]
- Kunzelmann, K. Ion channels in regulated cell death. Cell Mol. Life Sci. 2016, 73, 2387–2403. [Google Scholar] [CrossRef]
- Yun, J.; Lee, D.G. Role of potassium channels in chlorogenic acid-induced apoptotic volume decrease and cell cycle arrest in Candida albicans. Biochim. Biophys. Acta. 2017, 1861, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, D.G. Potential role of potassium and chloride channels in regulation of silymarin-induced apoptosis in Candida albicans. IUBMB Life 2018, 70, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Montague, J.W.; Bortner, C.D.; Hughes, F.M., Jr.; Cidlowski, J.A. A necessary role for reduced intracellular potassium during the DNA degradation phase of apoptosis. Steroids 1999, 649, 563–569. [Google Scholar] [CrossRef]
- Marklund, L.; Behnam-Motlagh, P.; Henriksson, R.; Grankvist, K. Bumetanide annihilation of amphotericin B-induced apoptosis and cytotoxicity is due to its effect on cellular K+ flux. J. Antimicrob. Chemother. 2001, 48, 781–786. [Google Scholar] [CrossRef]
- El Kebir, D.; József, L.; Khreiss, T.; Filep, J.G. Inhibition of K+ efflux prevents mitochondrial dysfunction, and suppresses caspase-3-, apoptosis-inducing factor-, and endonuclease G-mediated constitutive apoptosis in human neutrophils. Cell Signal. 2006, 18, 2302–2313. [Google Scholar] [CrossRef]
- Matsuyama, S.; Reed, J.C. Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ. 2000, 7, 1155–1165. [Google Scholar] [CrossRef]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar] [CrossRef]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef]
- Schüller, C.; Brewster, J.L.; Alexander, M.R.; Gustin, M.C.; Ruis, H. The HOG pathway controls osmotic regulation of transcription via the stress response element (STRE) of the Saccharomyces cerevisiae CTT1 gene. EMBO J. 1994, 13, 4382–4389. [Google Scholar] [CrossRef]
- Dechant, R.; Binda, M.; Lee, S.S.; Pelet, S.; Winderickx, J.; Peter, M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010, 29, 2515–2526. [Google Scholar] [CrossRef]
- Orij, R.; Brul, S.; Smits, G.J. Intracellular pH is a tightly controlled signal in yeast. Biochim. Biophys. Acta 2011, 1810, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Viejo-Diaz, M.; Andrés, M.T.; Fierro, J.F. Modulation of in vitro fungicidal activity of human lactoferrin against Candida albicans by extracellular cation concentration and target cell metabolic activity. Antimicrob. Agents Chemother. 2004, 48, 1242–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viejo-Diaz, M.; Andrés, M.T.; Fierro, J.F. Effects of human lactoferrin on the cytoplasmic membrane of Candida albicans cells related with its candidacidal activity. FEMS Immunol. Med. Microbiol. 2004, 42, 181–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergani, P.; Miosga, T.; Jarvis, S.M.; Blatt, M.R. Extracellular K+ and Ba2+ mediate voltage-dependent inactivation of the outward-rectifying K+ channel encoded by the yeast gene TOK1. FEBS Lett. 1997, 405, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Bertl, A.; Slayman, C.L.; Gradmann, D. Gating and conductance in an outward-rectifying K+ channel from the plasma membrane of Saccharomyces cerevisiae. J. Membr. Biol. 1993, 132, 183–199. [Google Scholar] [CrossRef]
- Baev, D.; Rivetta, A.; Li, X.S.; Vylkova, S.; Bashi, E.; Slayman, C.L.; Edgerton, M. Killing of Candida albicans by human salivary histatin 5 is modulated, but not determined, by the potassium channel TOK1. Infect. Immun. 2003, 71, 3251–3260. [Google Scholar] [CrossRef] [Green Version]
- Bañuelos, M.A.; Sychrová, H.; Bleykasten-Grosshans, C.; Souciet, J.L.; Potier, S. The Nhal antiporter of Saccharomyces cerevisiae mediates sodium and potassium efflux. Microbiology 1998, 144, 2749–2758. [Google Scholar]
- Marešová, L.; Hušeková, B.; Urbánková, E.; Chaloupka, R.; Sychrová, H. New applications of pHluorin-measuring intracellular pH of prototrophic yeasts and determining changes in the buffering capacity of strains with affected potassium homeostasis. Yeast 2010, 27, 317–325. [Google Scholar]
- Sokolov, S.; Knorre, D.; Smirnova, E.; Markova, O.; Pozniakovsky, A.; Skulachev, V.; Severin, F. Ysp2 mediates death of yeast induced by amiodarone or intracellular acidification. Biochim. Biophys. Acta 2006, 1757, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Lauff, D.B.; Santa-María, G.E. Potassium deprivation is sufficient to induce a cell death program in Saccharomyces cerevisiae. FEMS Yeast Res. 2010, 10, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedűs, N.; Leiter, E.; Kovács, B.; Tomori, V.; Kwon, N.-J.; Emri, T.; Marx, F.; Batta, G.; Csernoch, L.; Haas, H.; et al. The small molecular mass antifungal protein of Penicillium chrysogenum–a mechanism of action oriented review. J. Basic Microbiol. 2011, 51, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, M.; Nebe-von-Caron, G.; Steels, H.; Novodvorska, M.; Ueckert, J.; Archer, D.B. Weak-acid preservatives: pH and proton movements in the yeast Saccharomyces cerevisiae. Int. J. Food Microbiol. 2013, 161, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Yenush, L. Potassium and sodium transport in yeast. Adv. Exp. Med. Biol. 2016, 892, 187–228. [Google Scholar] [PubMed]
- Bertl, A.; Bihler, H.; Reid, J.D.; Kettner, C.; Slayman, C.L. Physiological characterization of the yeast plasma membrane outward rectifying K+ channel, DUK1 (TOK1), in situ. J. Membr. Biol. 1998, 162, 67–80. [Google Scholar] [CrossRef]
- Sousa, M.J.; Ludovico, P.; Rodrigues, F.; Leão, C.; Côrte-Real, M. Stress and cell death in yeast induced by acetic acid. In Cell Metabolism-Cell Homeostasis and Stress Response; Bubulya, P., Ed.; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef] [Green Version]
- Lastauskienė, E.; Zinkevičienė, A.; Girkontaitė, I.; Kaunietis, A.; Kvedarienė, V. Formic acid and acetic acid induce a programmed cell death in pathogenic Candida species. Curr. Microbiol. 2014, 69, 303–310. [Google Scholar] [CrossRef]
- Balzan, R.; Sapienza, K.; Galea, D.R.; Vassallo, N.; Frey, H.; Banister, W.H. Aspirin commits yeast cells to apoptosis depending on carbon source. Microbiology 2004, 150, 109–115. [Google Scholar] [CrossRef]
- Du, L.; Su, Y.; Sun, D.; Zhu, W.; Wang, J.; Zhuang, X.; Zhou, S.; Lu, Y. Formic acid induces Yca1p-independent apoptosis-like cell death in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 531–539. [Google Scholar] [CrossRef]
- Mutoh, N.; Kitajima, S.; Ichihara, S. Apoptotic cell death in the fission yeast Schizosaccharomyces pombe induced by valproic acid and its extreme susceptibility to pH change. Biosci. Biotechnol. Biochem. 2011, 75, 1113–1118. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Lee, D.G. A novel fungal killing mechanism of propionic acid. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef]
- Fonzi, W.A.; Irwin, M.Y. Isogenic strain construction and gene mapping in Candida albicans. Genetics 1993, 134, 717–728. [Google Scholar] [PubMed]
- Liu, N.-N.; Köhler, J.R. Antagonism of fluconazol and a proton pump inhibitor against Candida albicans. Antimicrob. Agents Chemother. 2016, 60, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.-N.; Flanagan, P.R.; Zeng, J.; Niketa, M.J.; Cardenas, M.E.; Moran, G.P.; Köhler, J.R. Phosphate is the third nutrient monitored by TOR in Candida albicans and provides a target for fungal-specific indirect TOR inhibition. Proc. Natl. Acad. Sci. USA 2017, 114, 6346–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, R. Effect of ATPase inhibitors on the proton pump of respiratory-deficient yeast. Eur. J. Biochem. 1980, 105, 419–424. [Google Scholar] [CrossRef]
- Diakov, T.T.; Tarsio, M.; Kane, P.M. Measurement of vacuolar and cytosolic pH in vivo in yeast cell suspensions. J. Vis. Exp. 2013, 74, e50261. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Ref. |
|---|---|---|
| ATCC 10231 | Wild type—clinical strain | ATCC |
| CAI4 | ura3::imm434/ura3::imm434 iro1/iro1::imm434TOK1/TOK1 | [52] |
| DBT2 | Δura3::imm434/Δura3::imm434 Δtok1/TOK1 | [37] |
| DBT3 | Δura3::imm434/Δura3::imm434 Δtok1/Δtok1 | [37] |
| JKC1562 | ACT1/promoterACT1-pHluorin-NAT1-ACT1his1/his1::tetR-FRT arg4/arg4 IRO1/iro1Δ::λimm434 URA3/ura3Δ::λimm434 | [53] |
| JKC1594 | ACT1/promoterACT1-NAT1-terminatorACT1his1/his1::tetR-FRT arg4/arg4 IRO1/iro1Δ::λimm434 URA3/ura3Δ::λimm434 | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, M.T.; Acosta-Zaldívar, M.; González-Seisdedos, J.; Fierro, J.F. Cytosolic Acidification Is the First Transduction Signal of Lactoferrin-Induced Regulated Cell Death Pathway. Int. J. Mol. Sci. 2019, 20, 5838. https://doi.org/10.3390/ijms20235838
Andrés MT, Acosta-Zaldívar M, González-Seisdedos J, Fierro JF. Cytosolic Acidification Is the First Transduction Signal of Lactoferrin-Induced Regulated Cell Death Pathway. International Journal of Molecular Sciences. 2019; 20(23):5838. https://doi.org/10.3390/ijms20235838
Chicago/Turabian StyleAndrés, María T., Maikel Acosta-Zaldívar, Jessica González-Seisdedos, and José F. Fierro. 2019. "Cytosolic Acidification Is the First Transduction Signal of Lactoferrin-Induced Regulated Cell Death Pathway" International Journal of Molecular Sciences 20, no. 23: 5838. https://doi.org/10.3390/ijms20235838
APA StyleAndrés, M. T., Acosta-Zaldívar, M., González-Seisdedos, J., & Fierro, J. F. (2019). Cytosolic Acidification Is the First Transduction Signal of Lactoferrin-Induced Regulated Cell Death Pathway. International Journal of Molecular Sciences, 20(23), 5838. https://doi.org/10.3390/ijms20235838