Specificity of Escherichia coli Heat-Labile Enterotoxin Investigated by Single-Site Mutagenesis and Crystallography

 and
and
Abstract
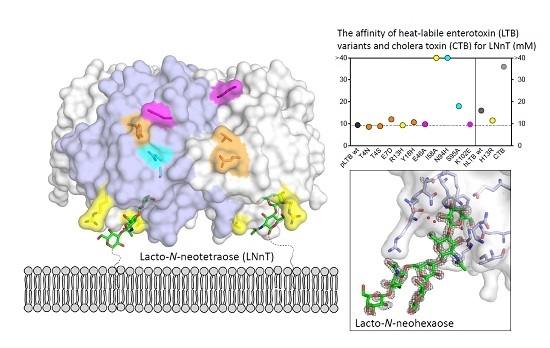
:
1. Introduction
2. Results
2.1. Quality Control
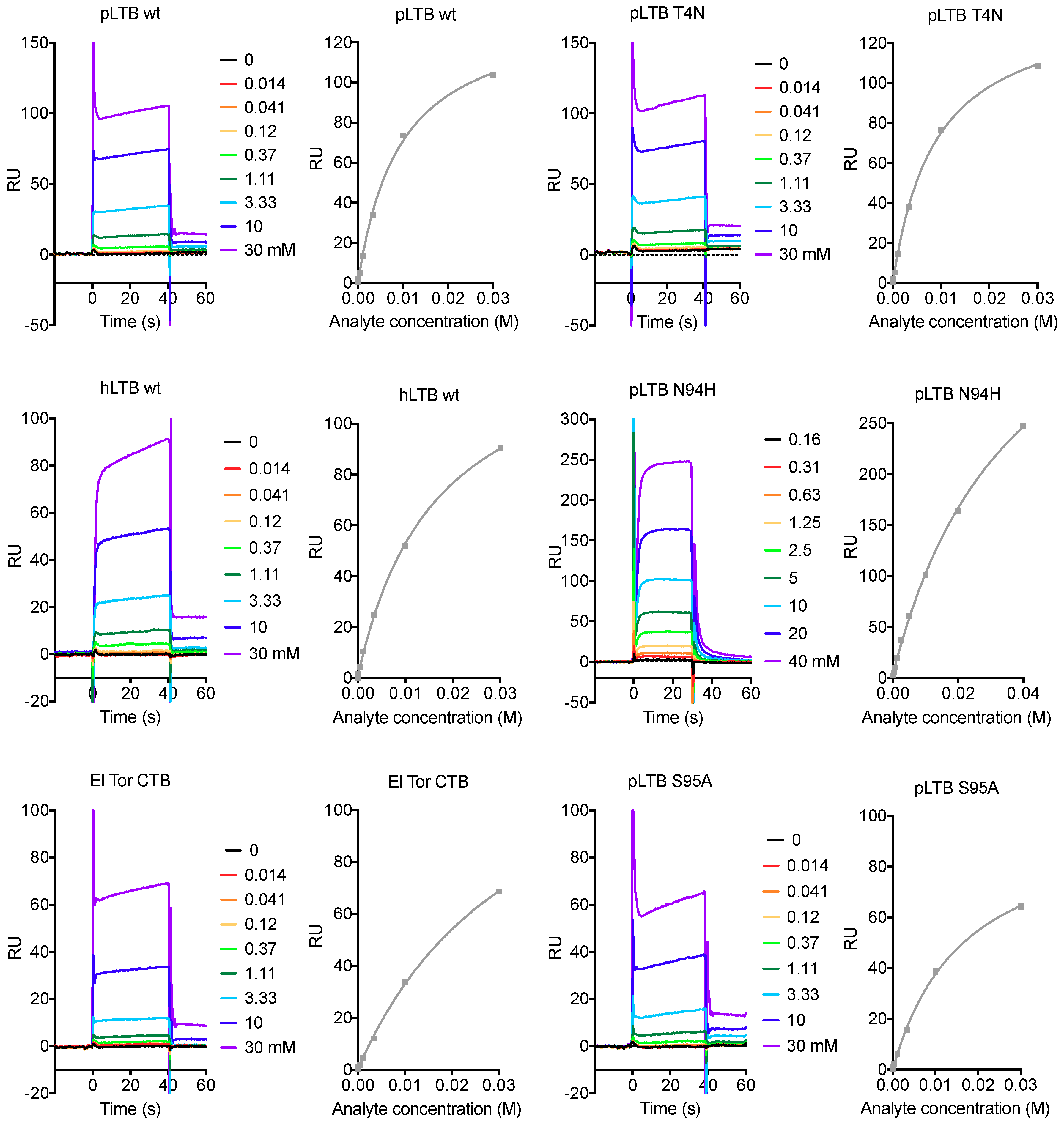
2.2. Surface Plasmon Resonance Spectroscopy with Lacto-N-Neotetraose
2.2.1. Primary Binding Site Residues 13 and 58
2.2.2. Residues 94 and 95
2.2.3. Secondary Binding Site Residues 4, 7, and 18
2.2.4. pLTB-Specific Residues 46 and 102
2.3. Surface Plasmon Resonance Spectroscopy with Lacto-N-Neohexaose
2.4. Crystal Structures of pLTB wt with Lacto-N-Neohexaose
3. Discussion
4. Materials and Methods
4.1. Generation of Single-Site Variants
4.2. Production and Purification of Protein
4.3. Analysis by Surface Plasmon Resonance
4.4. Crystallographic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| CD | circular dichroism |
| Cer | ceramide |
| CT | cholera toxin |
| CTB | cholera toxin, pentamer of B-subunits |
| GD2 | disialoganglioside GalNAcβ4[NeuAcα8NeuAcα3]Galβ4GlcβCer |
| GlcNAc | 2’-N-acetyl glucosamine |
| GM1 | monosialoganglioside Galβ3GalNAcβ4[NeuAcα3]Galβ4GlcβCer |
| hLT | Escherichia coli heat-labile enterotoxin from human isolates |
| hLTB | E. coli heat-labile enterotoxin from human isolates, pentamer of B-subunits |
| LNnH | lacto-N-neohexaose Galβ4GlcNAcβ6[Galβ4GlcNAcβ3]Galβ4Glc |
| LNnH-Cer | ganglioside neolactohexaosylceramide Galβ4GlcNAcβ6[Galβ4GlcNAcβ3]Galβ4GlcβCer |
| LNnT | lacto-N-neotetraose Galβ4GlcNAcβ3Galβ4Glc |
| LNnT-Cer | ganglioside neolactotetraosylceramide Galβ4GlcNAcβ3Galβ4GlcβCer (paragloboside) |
| LT | E. coli heat-labile enterotoxin |
| LTB | E. coli heat-labile enterotoxin, pentamer of B-subunits |
| NeuAc | 5’-N-acetyl neuraminic acid |
| PBS | phosphate-buffered saline |
| PDB | Protein Data Bank |
| pLT | E. coli heat-labile enterotoxin from porcine isolates |
| pLTB | E. coli heat-labile enterotoxin from porcine isolates, pentamer of B-subunits |
| r.m.s.d. | root mean square deviation |
| SPR | surface plasmon resonance |
References
- Khalil, I.A.; Troeger, C.; Blacker, B.F.; Rao, P.C.; Brown, A.; Atherly, D.E.; Brewer, T.G.; Engmann, C.M.; Houpt, E.R.; Kang, G.; et al. Morbidity and mortality due to shigella and enterotoxigenic Escherichia coli diarrhoea: The Global Burden of Disease Study 1990–2016. Lancet Infect. Dis. 2018, 11, 1229–1240. [Google Scholar] [CrossRef]
- Guerrant, R.L.; DeBoer, M.D.; Moore, S.R.; Scharf, R.J.; Lima, A.A.M. The impoverished gut−a triple burden of diarrhoea, stunting and chronic disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Halvorson, H.A.; Schlett, C.D.; Riddle, M.S. Postinfectious irritable bowel syndrome—A meta-analysis. Am. J. Gastroenterol. 2006, 101, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Hol, W.G.J. AB5 toxins. Curr. Opin. Struct. Biol. 1995, 5, 165–171. [Google Scholar] [CrossRef]
- Heggelund, J.E.; Bjørnestad, V.A.; Krengel, U. Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J.E., Ladant, D., Popoff, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 195–229. [Google Scholar] [CrossRef]
- Kuziemko, G.M.; Stroh, M.; Stevens, R.C. Cholera toxin binding affinity and specificity for gangliosides determined by surface plasmon resonance. Biochemistry 1996, 35, 6375–6384. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, W.B.; Precious, B.L.; Homans, S.W. Dissecting the cholera toxin−ganglioside GM1 interaction by isothermal titration calorimetry. J. Am. Chem. Soc. 2004, 126, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Lauer, S.; Goldstein, B.; Nolan, R.L.; Nolan, J.P. Analysis of cholera toxin−ganglioside interactions by flow cytometry. Biochemistry 2002, 41, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Kuhn, P.; Sarfaty, S.; Erbe, J.L.; Holmes, R.K.; Hol, W.G.J. The 1.25 Å resolution refinement of the cholera toxin B-pentamer: Evidence of peptide backbone strain at the receptor-binding site. J. Mol. Biol. 1998, 282, 1043–1059. [Google Scholar] [CrossRef]
- Holmner, Å.; Mackenzie, A.; Ökvist, M.; Jansson, L.; Lebens, M.; Teneberg, S.; Krengel, U. Crystal structures exploring the origins of the broader specificity of Escherichia coli heat-labile enterotoxin compared to cholera toxin. J. Mol. Biol. 2011, 406, 387–402. [Google Scholar] [CrossRef]
- Merritt, E.A.; Sarfaty, S.; Jobling, M.G.; Chang, T.; Holmes, R.K.; Hirst, T.R.; Hol, W.G.J. Structural studies of receptor binding by cholera toxin mutants. Protein Sci. 1997, 6, 1516–1528. [Google Scholar] [CrossRef] [Green Version]
- Fukuta, S.; Magnani, J.L.; Twiddy, E.M.; Holmes, R.K.; Ginsburg, V. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect. Immunity 1988, 56, 1748–1753. [Google Scholar]
- Ångström, J.; Teneberg, S.; Karlsson, K.A. Delineation and comparison of ganglioside-binding epitopes for the toxins of Vibrio cholerae, Escherichia coli, and Clostridium tetani: Evidence for overlapping epitopes. Proc. Natl. Acad. Sci. USA 1994, 91, 11859–11863. [Google Scholar] [CrossRef] [PubMed]
- Bäckström, M.; Shahabi, V.; Johansson, S.; Teneberg, S.; Kjellberg, A.; Miller-Podraza, H.; Holmgren, J.; Lebens, M. Structural basis for differential receptor binding of cholera and Escherichia coli heat-labile toxins: Influence of heterologous amino acid substitutions in the cholera B-subunit. Mol. Microbiol. 1997, 24, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Jansson, L.; Ångström, J.; Lebens, M.; Imberty, A.; Varrot, A.; Teneberg, S. Carbohydrate binding specificities and crystal structure of the cholera toxin-like B-subunit from Citrobacter freundii. Biochimie 2010, 92, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Teneberg, S.; Berntsson, A.; Ångström, J. Common architecture of the primary galactose binding sites of Erythrina corallodendron lectin and heat-labile enterotoxin from Escherichia coli in relation to the binding of branched neolactohexaosylceramide. J. Biochem. 2000, 128, 481–491. [Google Scholar] [CrossRef]
- Ångström, J.; Bäckström, M.; Berntsson, A.; Karlsson, N.; Holmgren, J.; Karlsson, K.-A.; Lebens, M.; Teneberg, S. Novel carbohydrate binding site recognizing blood group A and B determinants in a hybrid of cholera toxin and Escherichia coli heat-labile enterotoxin B-subunits. J. Biol. Chem. 2000, 275, 3231–3238. [Google Scholar] [CrossRef]
- Holmner, Å.; Lebens, M.; Teneberg, S.; Ångström, J.; Ökvist, M.; Krengel, U. Novel binding site identified in a hybrid between cholera toxin and heat-labile enterotoxin: 1.9 Å crystal structure reveals the details. Structure 2004, 12, 1655–1667, Erratum in: Structure 2007, 15, 253. [Google Scholar] [CrossRef]
- Holmner, Å.; Askarieh, G.; Ökvist, M.; Krengel, U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J. Mol. Biol. 2007, 371, 754–764. [Google Scholar] [CrossRef]
- Heggelund, J.E.; Burschowsky, D.; Bjørnestad, V.A.; Hodnik, V.; Anderluh, G.; Krengel, U. High-resolution crystal structures elucidate the molecular basis of cholera blood group dependence. PLoS Pathog. 2016, 12, e1005567. [Google Scholar] [CrossRef]
- Heggelund, J.E.; Haugen, E.; Lygren, B.; Mackenzie, A.; Holmner, Å.; Vasile, F.; Reina, J.J.; Bernardi, A.; Krengel, U. Both El Tor and classical cholera toxin bind blood group determinants. Biochem. Biophys. Res. Commun. 2012, 418, 731–735. [Google Scholar] [CrossRef]
- Mandal, P.K.; Branson, T.R.; Hayes, E.D.; Ross, J.F.; Gavín, J.A.; Daranas, A.H.; Turnbull, W.B. Towards a structural basis for the relationship between blood group and the severity of El Tor cholera. Angew. Chem. Int. Ed. Engl. 2012, 51, 5143–5146. [Google Scholar] [CrossRef]
- Vasile, F.; Reina, J.J.; Potenza, D.; Heggelund, J.E.; Mackenzie, A.; Krengel, U.; Bernardi, A. Comprehensive analysis of blood group antigen binding to classical and El Tor cholera toxin B-pentamers by NMR. Glycobiology 2014, 24, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmner-Rocklöv, Å. Molecular recognition of carbohydrates—Structural and functional Characterisation of bacterial toxins and fungal lectins. Ph.D. Thesis, Chalmers University of Technology, Gothenburg, Sweden, 2005. [Google Scholar]
- Jansson, L.; Ångström, J.; Lebens, M.; Teneberg, S. No direct binding of the heat-labile enterotoxin of Escherichia coli to E. coli lipopolysaccharides. Glycoconj. J. 2010, 27, 171–179. [Google Scholar] [CrossRef]
- Heim, J.B.; Hodnik, V.; Heggelund, J.E.; Anderluh, G.; Krengel, U. Crystal structures reveal that Lewis-x and fucose bind to secondary cholera toxin binding site-in contrast to fucosyl-GM1. bioRxiv 2018, 2018, 431130. [Google Scholar] [CrossRef]
- Hatlem, D.; Heggelund, J.E.; Burschowsky, D.; Krengel, U.; Kristiansen, P.E. 1H, 13C, 15N backbone assignment of the human heat-labile enterotoxin B-pentamer and chemical shift mapping of neolactotetraose binding. Biomol. NMR Assign. 2017, 11, 99–104. [Google Scholar] [CrossRef]
- Karlsson, K.-A.; Teneberg, S.; Ångström, J.; Kjellberg, A.; Hirst, T.R.; Bergström, J.; Miller-Podraza, H. Unexpected carbohydrate cross-binding by Escherichia coli heat-labile enterotoxin. Recognition of human and rabbit target cell glycoconjugates in comparison with cholera toxin. Bioorg. Med. Chem. 1996, 4, 1919–1928. [Google Scholar] [CrossRef]
- Heggelund, J.E.; Mackenzie, A.; Martinsen, T.; Heim, J.B.; Cheshev, P.; Bernardi, A.; Krengel, U. Towards new cholera prophylactics and treatment: Crystal structures of bacterial enterotoxins in complex with GM1 mimics. Sci. Rep. 2017, 7, 2326. [Google Scholar] [CrossRef]
- Lebens, M.; Shahabi, V.; Bäckström, M.; Houze, T.; Lindblad, N.; Holmgren, J. Synthesis of hybrid molecules between heat-labile enterotoxin and cholera toxin B subunits: Potential for use in a broad-spectrum vaccine. Infect. Immunity 1996, 64, 2144–2150. [Google Scholar]
- Harris, J.B. Cholera: Immunity and prospects in vaccine development. J. Infect. Dis. 2018, 218, S141–S146. [Google Scholar] [CrossRef]
- Francis, M.L.; Ryan, J.; Jobling, M.G.; Holmes, R.K.; Moss, J.; Mond, J.J. Cyclic AMP-independent effects of cholera toxin on B cell activation. II. Binding of ganglioside GM1 induces B cell activation. J. Immunol. 1992, 148, 1999–2005. [Google Scholar]
- Nashar, T.O.; Webb, H.M.; Eaglestone, S.; Williams, N.A.; Hirst, T.R. Potent immunogenicity of the B subunits of Escherichia coli heat-labile enterotoxin: Receptor binding is essential and induces differential modulation of lymphocyte subsets. Proc. Natl. Acad. Sci. USA 1996, 93, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, J.; Bourgeois, L.; Carlin, N.; Clements, J.; Gustafsson, B.; Lundgren, A.; Nygren, E.; Tobias, J.; Walker, R.; Svennerholm, A.-M. Development and preclinical evaluation of safety and immunogenicity of an oral ETEC vaccine containing inactivated E. coli bacteria overexpressing colonization factors CFA/I, CS3, CS5 and CS6 combined with a hybrid LT/CT B subunit antigen, administered alone and together with dmLT adjuvant. Vaccine 2013, 31, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Chowdhury, M.I.; Bhuiyan, T.R.; Kaim, J.; Ahmed, T.; Rafique, T.A.; Khan, A.; Rahman, S.I.A.; Khanam, F.; Begum, Y.A.; et al. Evaluation of the safety and immunogenicity of the oral inactivated multivalent enterotoxigenic Escherichia coli vaccine ETVAX in Bangladeshi adults in a double-blind, randomized, placebo-controlled Phase I trial using electrochemiluminescence and ELISA assays for immunogenicity analyses. Vaccine 2018. [Google Scholar] [CrossRef]
- Norton, E.B.; Lawson, L.B.; Freytag, L.C.; Clements, J.D. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 2011, 18, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Potterton, L.; Agirre, J.; Ballard, C.; Cowtan, K.; Dodson, E.; Evans, P.R.; Jenkins, H.T.; Keegan, R.; Krissinel, E.; Stevenson, K.; et al. CCP4i2: The new graphical user interface to the CCP4 program suite. Acta Crystallogr. D Struct. Biol. 2018, 74, 68–84. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Minke, W.E.; Pickens, J.C.; Merritt, E.A.; Fan, E.; Verlinde, C.L.M.J.; Hol, W.G.J. Structure of m-carboxyphenyl-α-d-galactopyranoside complexed to heat-labile enterotoxin at 1.3 Å resolution: Surprising variations in ligand-binding modes. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 795–804. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.-W.; Jain, S.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Nicholls, R.A.; Emsley, P.; Gražulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A stereochemical description generator for ligands. Acta Crystallogr. D Struct. Biol. 2017, 73, 112–122. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
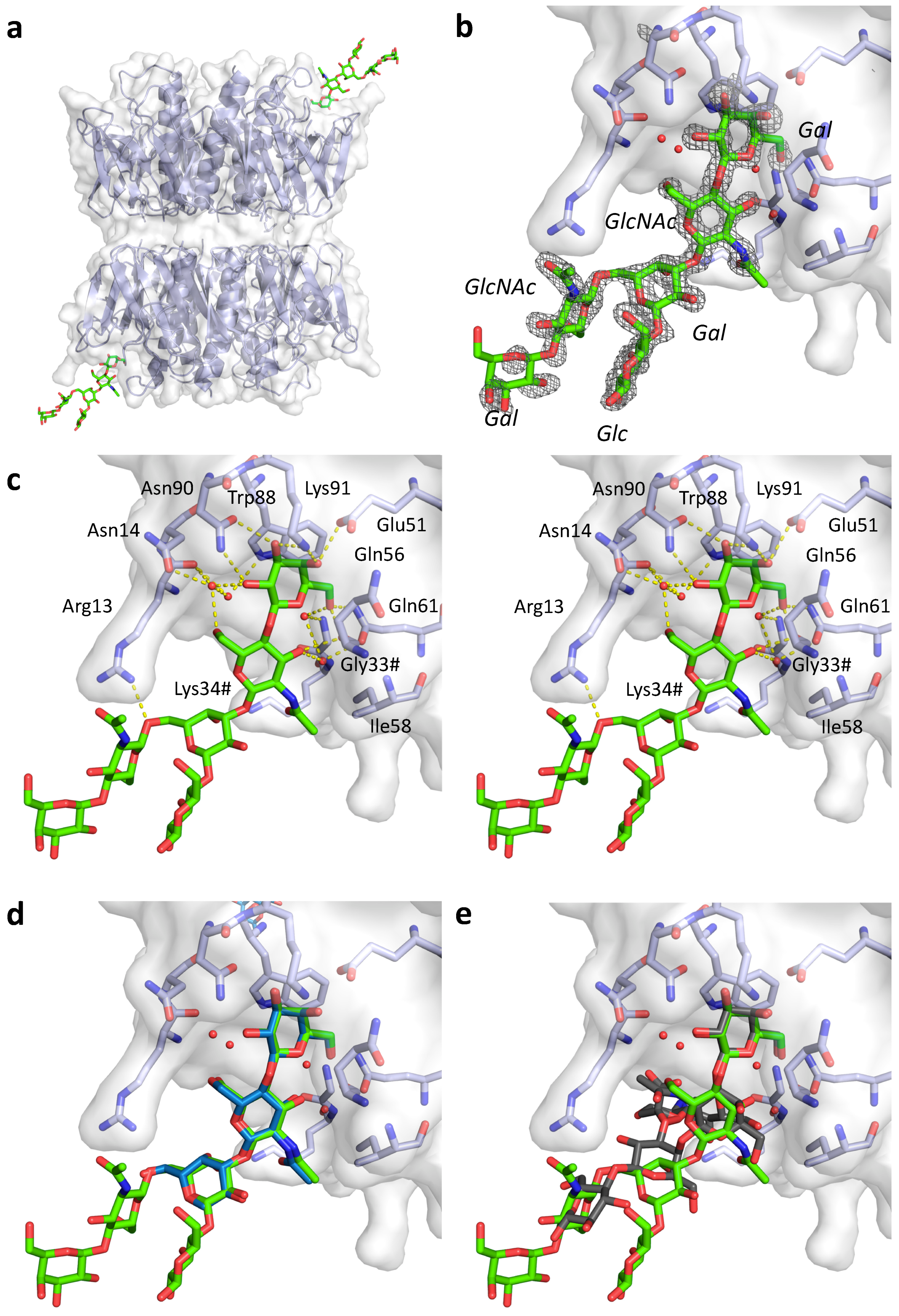{kind=link}
| Protein Variant | GM1a | LNnT Batch 1 | LNnT Batch 2 | LNnT Batch 3 | LNnT Batch 4 | LNnH |
|---|---|---|---|---|---|---|
| pLTB wt | 37 ± 0.5 nM | 6.7 ± 0.5 mM | - | 9.4 ± 0.1 mM | 8.0 ± 0.6 mM | 5 ± 1 mM |
| pLTB T4N | 32 ± 0.3 nM | - | - | 8.6 ± 0.1 mM | 7.8 ± 0.7 mM | - |
| pLTB T4S | - | - | - | 8.9 ± 0.4 mM | - | - |
| pLTB E7D | - | - | 12 ± 3 mM * | - | - | - |
| pLTB R13H | - | 9.3 ± 1.7 mM | - | - | - | 7 ± 1 mM |
| hLTB H13R | - | 11.5 ± 0.2 mM | - | - | - | - |
| pLTB Y18H | - | - | - | 10.7 ± 0.1 mM | - | - |
| pLTB E46A | - | - | - | 9.8 ± 0.1 mM | - | - |
| pLTB I58A | 2.3 ± 1.7 μM | - | n.b. | - | - | - |
| pLTB N94H | 23 ± 1 nM | - | >40 mM ** | - | - | - |
| pLTB S95A | 65 ± 4 nM | - | - | 18 ± 1.4 mM | - | - |
| pLTB K102E | 34 ± 0.8 nM | - | - | 9.7 ± 0.5 mM | - | - |
| hLTB wt | - | - | - | 16 ± 0.03 mM | - | - |
| CTB | - | - | - | >36 mM ** | - | - |
| Residue | pLTB | hLTB | CTB * | Toxin Variant | Effect on LNnT Affinity |
|---|---|---|---|---|---|
| 1 | Ala | Ala | Thr * | ||
| 4 ** | Thr | Ser | Asn | pLTB T4N/T4S | Like pLTB wt |
| 7 | Glu | Glu | Asp | pLTB E7D | Lower affinity |
| 10 | Ser | Ser | Ala | - | - |
| 13 | Arg | His | His | pLTB R13H | Similar to pLTB wt |
| hLTB H13R | Similar to hLTB wt | ||||
| 18 | Tyr | Tyr | Tyr * | pLTB Y18H | Slightly lower affinity |
| 20 | Ile | Ile | Leu * | - | - |
| 25 | Leu | Leu | Phe | - | - |
| 31 | Met | Met | Leu | - | - |
| 38 | Val | Val | Ala | - | - |
| 44 | Ser | Ser | Asn | - | - |
| 46 | Glu | Ala | Ala | pLTB E46A | Like pLTB wt |
| 47 | Thr | Thr | Ile * | - | - |
| 58 | Ile | Ile | Ile | pLTB I58A | No binding |
| 75 | Thr | Thr | Ala | - | - |
| 80 | Thr | Thr | Ala | - | - |
| 82 | Ile | Ile | Val | - | - |
| 83 | Asp | Asp | Glu | - | - |
| 94 | Asn | Asn | His | pLTB N94H | Lower affinity |
| 95 | Ser | Ser | Ala | pLTB S95A | Lower affinity |
| 102 | Lys | Glu | Ala | pLTB K102E | Like pLTB wt |
| Protein | pLTB + LNnH |
|---|---|
| PDB ID | 6IAL |
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 77.1, 65.6, 96.3 |
| β (°) | 108.6 |
| Resolution (Å) | 68.7–1.45 * (1.47–1.45) ** |
| No. of unique reflections | 160,263 (7,848) |
| CC(1/2) (%) | 99.6 (45.8) |
| Rmerge | 0.13 (1.15) |
| (I)/σ(I) | 6.4 (1.3) |
| Multiplicity | 4.4 (4.5) |
| Completeness (%) | 99.6 (99.0) |
| Refinement | |
| Rcryst/Rfree (%) | 17.5/20.1 |
| No. of atoms | |
| Protein | 8622 |
| Ligand/ion | 158/15 |
| Water | 644 |
| Average B-factors (Å2) | |
| Protein | 19.6 |
| Ligand /ion | 23.2/19.7 |
| Water | 23.2 |
| r.m.s.d. bonds (Å) | 0.01 |
| r.m.s.d. angles (°) | 1.7 |
| Residue | Donor/acceptor | Distance (Å) Site 1 | Distance (Å) Site 2 |
|---|---|---|---|
| Arg13 | NH1-O6 Galβ4 (second Gal) | 2.9 | 3.2 |
| Asn14 | OD1-O2 Galβ3 via solvent | 3.0–H2O–2.9 | - |
| and O6 GlcNAcβ3 via solvent | 3.0–H2O–2.8 | - | |
| Gly33# | N-O6 Galβ4 via solvent | 2.8–H2O–2.9 | 2.9–H2O–3.0 |
| Glu51 | OE2-O4 Galβ4 | 2.7 | 2.7 |
| Gln56 | O-O6 Galβ4 | 2.7 | 2.6 |
| O-O3 GlcNAcβ3 | 2.8 | 2.9 | |
| Ile58 | GlcNAcβ3 | 3.6* | 4.3 * |
| Gln61 | NE2-O6 Galβ4 | 3.0 | 3.0 |
| OD1-O3 GlcNAcβ3 via solvent | 2.8 – H2O – 2.8 | 2.9–H2O–2.9 | |
| Asn90 | ND2-O2 Galβ4 | 2.9 | 2.9 |
| OD1-O3 Galβ4 | 2.9 | 2.9 | |
| Lys91 | NZ-O3 Galβ4 | 2.8 | 2.8 |
| NZ-O4 Galβ4 | 2.9 | 2.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heggelund, J.E.; Heim, J.B.; Bajc, G.; Hodnik, V.; Anderluh, G.; Krengel, U. Specificity of Escherichia coli Heat-Labile Enterotoxin Investigated by Single-Site Mutagenesis and Crystallography. Int. J. Mol. Sci. 2019, 20, 703. https://doi.org/10.3390/ijms20030703
Heggelund JE, Heim JB, Bajc G, Hodnik V, Anderluh G, Krengel U. Specificity of Escherichia coli Heat-Labile Enterotoxin Investigated by Single-Site Mutagenesis and Crystallography. International Journal of Molecular Sciences. 2019; 20(3):703. https://doi.org/10.3390/ijms20030703
Chicago/Turabian StyleHeggelund, Julie Elisabeth, Joel Benjamin Heim, Gregor Bajc, Vesna Hodnik, Gregor Anderluh, and Ute Krengel. 2019. "Specificity of Escherichia coli Heat-Labile Enterotoxin Investigated by Single-Site Mutagenesis and Crystallography" International Journal of Molecular Sciences 20, no. 3: 703. https://doi.org/10.3390/ijms20030703
APA StyleHeggelund, J. E., Heim, J. B., Bajc, G., Hodnik, V., Anderluh, G., & Krengel, U. (2019). Specificity of Escherichia coli Heat-Labile Enterotoxin Investigated by Single-Site Mutagenesis and Crystallography. International Journal of Molecular Sciences, 20(3), 703. https://doi.org/10.3390/ijms20030703