The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art



Abstract
:1. Introduction
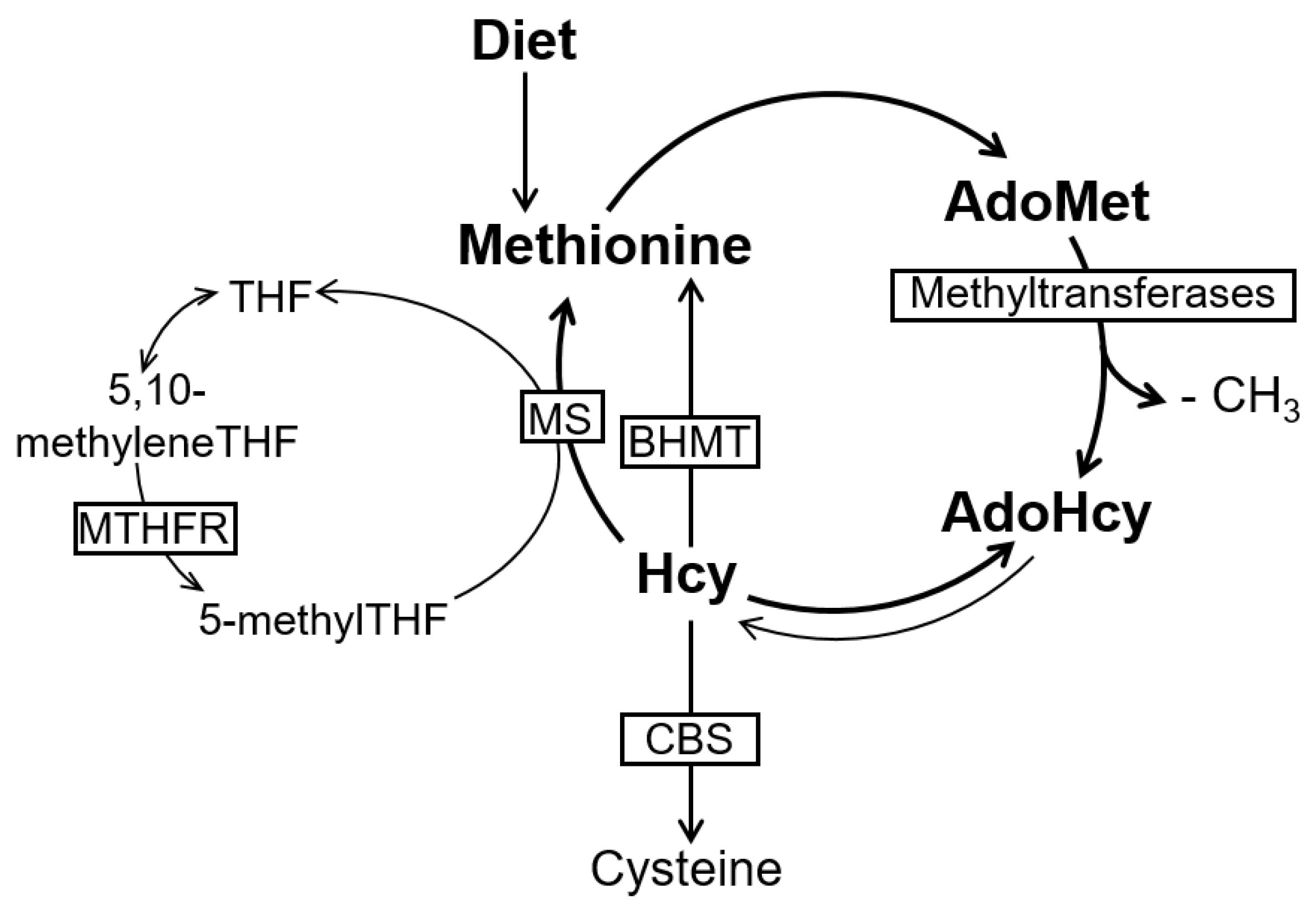
2. The Methionine Metabolism and Cell Methylation Status
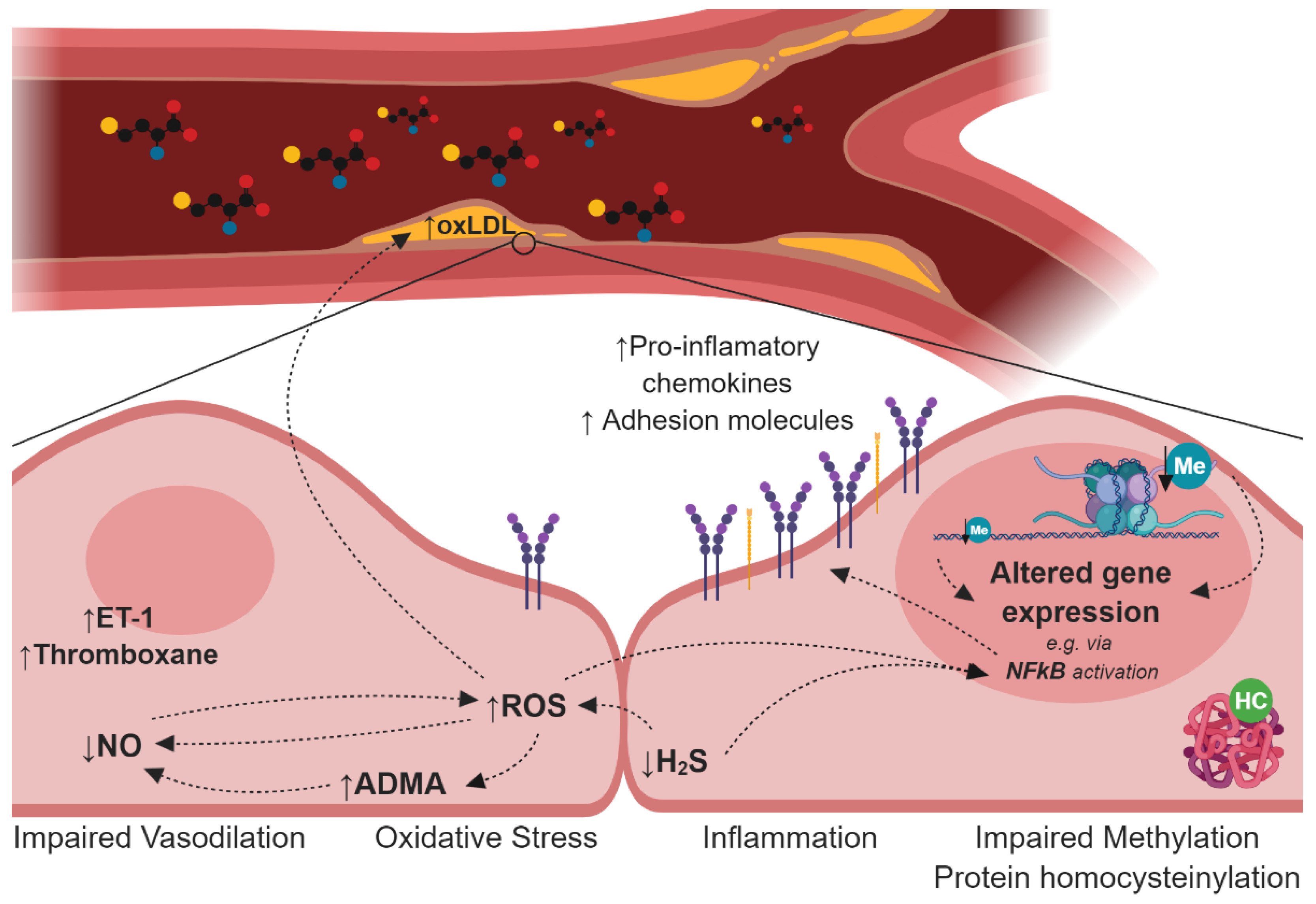
3. Endothelial Dysfunction and Atherosclerosis in Hyperhomocysteinemia (HHcy)
4. Mechanisms Induced by HHcy Associated with Endothelial Dysfunction and Atherogenesis
4.1. Impairment of the Nitric Oxide Synthesis
4.2. Deregulation of the Hydrogen Sulfide Signalling Pathway
4.3. Oxidative Stress
4.4. Disturbances in Lipoprotein Metabolism
4.5. Protein N-Homocysteinylation
4.6. Cellular Hypomethylation
5. Controversy Regarding the Negative Results from Hcy-Lowering Human Trials
6. Conclusions
Funding
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 October 2018).
- Stewart, J.; Manmathan, G.; Wilkinson, P. Primary prevention of cardiovascular disease: A review of contemporary guidance and literature. JRSM Cardiovasc. Dis. 2017, 6, 2048004016687211. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Handy, D.E.; Castro, R. The Link Between Hyperhomocysteinemia and Hypomethylation. J. Inborn Errors Metab. Screen. 2017, 5, 2326409817698994. [Google Scholar] [CrossRef]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.-G.; Banerjee, R. Homocysteine and redox signaling. Antioxid. Redox Signal. 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Schalinske, K.L.; Smazal, A.L. Homocysteine Imbalance: A Pathological Metabolic Marker. Adv. Nutr. Int. Rev. J. 2012, 3, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M. Pharmacological and biochemical aspects of S-adenosylhomocysteine and S-adenosylhomocysteine hydrolase. Pharmacol. Rev. 1982, 34, 223–253. [Google Scholar] [PubMed]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Weir, D.G.; Molloy, A.; McPartlin, J.; Daly, L.; Kirke, P. Folic acid metabolism and mechanisms of neural tube defects. Ciba Found. Symp. 1994, 181, 180–187. [Google Scholar]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Durand, P.; Prost, M.; Loreau, N.; Lussier-Cacan, S.; Blache, D. Impaired homocysteine metabolism and atherothrombotic disease. Lab. Investig. 2001, 81, 645–672. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Ueki, I. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011, 34, 17–32. [Google Scholar] [CrossRef]
- Taysi, S.; Keles, M.S.; Gumustekin, K.; Akyuz, M.; Boyuk, A.; Cikman, O.; Bakan, N. Plasma homocysteine and liver tissue S-adenosylmethionine, S-adenosylhomocysteine status in Vitamin B6-deficient rats. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 154–160. [Google Scholar]
- Finkelstein, J.D. The metabolism of homocysteine: Pathways and regulation. Eur. J. Pediatr. 1998, 157, S40–S44. [Google Scholar] [CrossRef]
- Mudd, S.H.; Cerone, R.; Schiaffino, M.C.; Fantasia, A.R.; Minniti, G.; Caruso, U.; Lorini, R.; Watkins, D.; Matiaszuk, N.; Rosenblatt, D.S.; et al. Glycine N-methyltransferase deficiency: A novel inborn error causing persistent isolated hypermethioninaemia. J. Inherit. Metab. Dis. 2001, 24, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D.; Martin, J.J. Methionine metabolism in mammals. Adaptation to methionine excess. J. Biol. Chem. 1986, 261, 1582–1587. [Google Scholar] [CrossRef]
- Watkins, D.; Rosenblatt, D.S.; Fowler, B. Disorders of cobalamin and folate transport and metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2012; pp. 385–402. ISBN 9783642157202. [Google Scholar]
- Blom, H.J. Consequences of homocysteine export and oxidation in the vascular system. Semin. Thromb. Hemost. 2000, 26, 227–232. [Google Scholar] [CrossRef]
- Sengupta, S.; Chen, H.; Togawa, T.; DiBello, P.M.; Majors, A.K.; Büdy, B.; Ketterer, M.E.; Jacobsen, D.W. Albumin Thiolate Anion Is an Intermediate in the Formation of Albumin-S-S-Homocysteine. J. Biol. Chem. 2001, 276, 30111–30117. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A.C.; Fung, E.; Bieda, M.C.; Snyder, F.F.; Gravel, R.A.; Cross, J.C.; et al. Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 2013, 155, 81–93. [Google Scholar] [CrossRef]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef]
- Čejková, S.; Králová-Lesná, I.; Poledne, R. Monocyte adhesion to the endothelium is an initial stage of atherosclerosis development. Cor Vasa 2016, 58, e419–e425. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.D. Homocysteine lowering for stroke prevention: Unravelling the complexity of the evidence. Int. J. Stroke 2016, 11, 744–747. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, C.; Zhou, Y.; Yan, T.; Wang, Z.; Li, W.; Zhao, L. Homocysteine Induces Apoptosis of Human Umbilical Vein Endothelial Cells via Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Barroso, M.; Kao, D.; Blom, H.J.; Tavares de Almeida, I.; Castro, R.; Loscalzo, J.; Handy, D.E. S-adenosylhomocysteine induces inflammation through NFkB: A possible role for EZH2 in endothelial cell activation. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Barroso, M.; Florindo, C.; Kalwa, H.; Silva, Z.; Turanov, A.A.; Carlson, B.A.; De Almeida, I.T.; Blom, H.J.; Gladyshev, V.N.; Hatfield, D.L.; et al. Inhibition of cellular methyltransferases promotes endothelial cell activation by suppressing glutathione peroxidase 1 protein expression. J. Biol. Chem. 2014, 289, 15350–15362. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, I.; Barroso, M.; Moura, T.; Castro, R.; Soveral, G. Endothelial Aquaporins and Hypomethylation: Potential Implications for Atherosclerosis and Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 130. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Rodionov, R.N.; Arning, E.; Bottiglieri, T.; Kimoto, M.; Murry, D.J.; Cooke, J.P.; Faraci, F.M.; Lentz, S.R. Tissue-specific downregulation of dimethylarginine dimethylaminohydrolase in hyperhomocysteinemia. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H816–H825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gottsäter, A.; Anwaar, I.; Eriksson, K.-F.; Mattiasson, I.; Lindgärde, F.; Gottsäter, A. Homocysteine Is Related to Neopterin and Endothelin-1 in Plasma of Subjects with Disturbed Glucose Metabolism and Reference Subjects. Angiology 2000, 51, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, G.; Davì, G.; Margaglione, M.; Cirillo, F.; Grandone, E.; Ciabattoni, G.; Catalano, I.; Strisciuglio, P.; Andria, G.; Patrono, C.; et al. Abnormally high thromboxane biosynthesis in homozygous homocystinuria. Evidence for platelet involvement and probucol-sensitive mechanism. J. Clin. Investig. 1993, 92, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef]
- Wald, D.S.; Morris, J.K.; Wald, N.J. Reconciling the Evidence on Serum Homocysteine and Ischaemic Heart Disease: A Meta-Analysis. PLoS ONE 2011, 6, e16473. [Google Scholar] [CrossRef]
- Yuyun, M.F.; Ng, L.L.; Ng, G.A. Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc. Res. 2018, 119, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Pierini, D.; Bryan, N.S. Nitric Oxide Availability as a Marker of Oxidative Stress. In Advanced Protocols in Oxidative Stress III; Humana Press: New York, NY, USA, 2015; pp. 63–71. [Google Scholar]
- Subelzu, N.; Bartesaghi, S.; de Bem, A.; Radi, R. Oxidative Inactivation of Nitric Oxide and Peroxynitrite Formation in the Vasculature. In Oxidative Stress: Diagnostics and Therapy; American Chemical Society: Washington, DC, USA, 2015; pp. 91–145. [Google Scholar]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Gielis, J.F.; Lin, J.Y.; Wingler, K.; Van Schil, P.E.Y.; Schmidt, H.H.; Moens, A.L. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic. Biol. Med. 2011, 50, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran, L.J.; Kohl, B.; Rao, V.; Kisiel, W.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiang, Y.; Yang, A.; Sun, W.; Ma, C.; Ma, S.; Gong, H.; Shi, Y.; Wei, J. Hyperhomocysteinemia-Induced Monocyte Chemoattractant Protein-1 Promoter DNA Methylation by Nuclear Factor-κB/DNA Methyltransferase 1 in Apolipoprotein E–Deficient Mice. Biores. Open Access 2013, 2, 118–127. [Google Scholar] [CrossRef]
- Sipkens, J.A.; Hahn, N.; den Brand, C.S.; Meischl, C.; Cillessen, S.A.G.M.; Smith, D.E.C.; Juffermans, L.J.M.; Musters, R.J.P.; Roos, D.; Jakobs, C.; et al. Homocysteine-Induced Apoptosis in Endothelial Cells Coincides With Nuclear NOX2 and Peri-nuclear NOX4 Activity. Cell Biochem. Biophys. 2013, 67, 341–352. [Google Scholar] [CrossRef]
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999, 43, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, H.; Matsuoka, H.; Cooke, J.P.; Usui, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Endogenous nitric oxide synthase inhibitor: A novel marker of atherosclerosis. Circulation 1999, 99, 1141–1146. [Google Scholar] [CrossRef]
- Nijveldt, R.J.; Teerlink, T.; van Leeuwen, P.A.M. The asymmetrical dimethylarginine (ADMA)-multiple organ failure hypothesis. Clin. Nutr. 2003, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Millatt, L.J.; Whitley, G.S.J.; Li, D.; Leiper, J.M.; Siragy, H.M.; Carey, R.M.; Johns, R.A. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation 2003, 108, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.H.; Lee, S.C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001, 158, 425–430. [Google Scholar] [CrossRef]
- Schnog, J.B.; Teerlink, T.; van der Dijs, F.P.L.; Duits, A.J.; Muskiet, F.A.J. Plasma levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell disease. Ann. Hematol. 2005, 84, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T. Determination of the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine in biological samples by HPLC. Methods Mol. Med. 2005, 108, 263–274. [Google Scholar] [PubMed]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Förstermann, U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide Biol. Chem. 1997, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Ogawa, T.; Cooke, J.P. Novel mechanism for endothelial dysfunction: Dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 1999, 99, 3092–3095. [Google Scholar] [CrossRef]
- Konishi, H.; Sydow, K.; Cooke, J.P. Dimethylarginine Dimethylaminohydrolase Promotes Endothelial Repair After Vascular Injury. J. Am. Coll. Cardiol. 2007, 49, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Sydow, K.; Gunawan, F.; Jacobi, J.; Tsao, P.S.; Robbins, R.C.; Cooke, J.P. Dimethylarginine dimethylaminohydrolase overexpression suppresses graft coronary artery disease. Circulation 2005, 112, 1549–1556. [Google Scholar] [CrossRef]
- Böger, R.H.; Bode-Böger, S.M.; Sydow, K.; Heistad, D.D.; Lentz, S.R. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in monkeys with hyperhomocyst(e)inemia or hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B.; Tsikas, D.; Bode-Böger, S.M. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: Involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 2000, 87, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.H.; Guo, Z.; Feng, M.; Wu, Z.Z.; He, Z.M.; Xiong, Y. Protection of DDAH2 overexpression against homocysteine-induced impairments of DDAH/ADMA/NOS/NO pathway in endothelial cells. Cell. Physiol. Biochem. 2012, 30, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef]
- Zhu, W.; Lin, A.; Banerjee, R. Kinetic properties of polymorphic variants and pathogenic mutants in human cystathionineγ-lyase. Biochemistry 2008, 47, 6226–6232. [Google Scholar] [CrossRef]
- Chen, X.; Jhee, K.H.; Kruger, W.D. Production of the neuromodulator H2S by cystathionine??-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem. 2004, 279, 52082–52086. [Google Scholar] [CrossRef]
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell. Mol. Med. 2009, 13, 488–507. [Google Scholar] [CrossRef]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Jhee, K.H.; McPhie, P.; Miles, E.W. Domain architecture of the heme-independent yeast cystathionine β -synthase provides insights into mechanisms of catalysis and regulation. Biochemistry 2000, 39, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Łowicka, E.; Bełtowski, J. Hydrogen sulfide (H2S)—The third gas of interest for pharmacologists. Pharmacol. Rep. 2007, 59, 4–24. [Google Scholar] [PubMed]
- Nandi, S.S.; Mishra, P.K. H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 2017, 7, 3639. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer. Molecules 2017, 22, 582. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen Sulfide and Endothelial Dysfunction: Relationship with Nitric Oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef]
- Zuidema, M.Y.; Peyton, K.J.; Fay, W.P.; Durante, W.; Korthuis, R.J. Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: Role of heme oxygenase-1. Am. J. Physiol. Circ. Physiol. 2011, 301, H888–H894. [Google Scholar] [CrossRef]
- Muzaffar, S.; Jeremy, J.Y.; Sparatore, A.; Del Soldato, P.; Angelini, G.D.; Shukla, N. H 2S-donating sildenafil (ACS6) inhibits superoxide formation and gp91 phox expression in arterial endothelial cells: Role of protein kinases A and G. Br. J. Pharmacol. 2008, 155, 984–994. [Google Scholar] [CrossRef]
- Suo, R.; Zhao, Z.Z.; Tang, Z.H.; Ren, Z.; Liu, X.; Liu, L.S.; Wang, Z.; Tang, C.K.; Wei, D.H.; Jiang, Z.S. Hydrogen sulfide prevents H2O2-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol. Med. Rep. 2013, 7, 1865–1870. [Google Scholar] [CrossRef]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.-L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine -Lyase Protects against Renal Ischemia/Reperfusion by Modulating Oxidative Stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S -Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.K.; Chang, T.; Wang, H.; Wu, L.; Wang, R.; Meng, Q.H. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Wang, H.; Kho, S.H.; Rinkiko, S.; Sheng, X.; Shen, H.M.; Zhu, Y.Z. Hydrogen Sulfide Protects HUVECs against Hydrogen Peroxide Induced Mitochondrial Dysfunction and Oxidative Stress. PLoS ONE 2013, 8, e53147. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Signaling pathways for the vascular effects of hydrogen sulfide. Curr. Opin. Nephrol. Hypertens. 2011, 20, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Szabo, C.; Ichinose, F.; Ahmed, A.; Whiteman, M.; Papapetropoulos, A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol. Sci. 2015, 36, 568–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Szabó, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, Š.; Meng, Q.H.; Wang, R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Jin, H.; Wei, H.; Li, W.; Bu, D.; Tang, X.; Ren, Y.; Tang, C.; Du, J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein e knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E-/-mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.H.; Kwok, W.K.; To, K.F.; Lau, J.Y.W. Anti-atherogenic effect of Hydrogen sulfide by over-expression of cystathionine gamma-lyase (CSE) gene. PLoS ONE 2014, 9, e113038. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X. Updating the relationship between hyperhomocysteinemia lowering therapy and cardiovascular events. Cardiovasc. Ther. 2013, 31, e19–e26. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial Dysfunction: The Link Between Homocysteine and Hydrogen Sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef]
- Xue, H.; Zhou, S.; Xiao, L.; Guo, Q.; Liu, S.; Wu, Y. Hydrogen sulfide improves the endothelial dysfunction in renovascular hypertensive rats. Physiol. Res. 2015, 64, 663–672. [Google Scholar]
- Zhao, Z.; Liu, X.; Shi, S.; Li, H.; Gao, F.; Zhong, X.; Wang, Y. Exogenous hydrogen sulfide protects from endothelial cell damage, platelet activation, and neutrophils extracellular traps formation in hyperhomocysteinemia rats. Exp. Cell Res. 2018, 370, 434–443. [Google Scholar] [CrossRef]
- Saha, S.; Chakraborty, P.K.; Xiong, X.; Dwivedi, S.K.D.; Mustafi, S.B.; Leigh, N.R.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016, 30, 441–456. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Yadav, V.; Banerjee, R. Heme-dependent metabolite switching regulates H2S synthesis in response to Endoplasmic Reticulum (ER) stress. J. Biol. Chem. 2016, 291, 16418–16423. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Faverzani, J.L.; Hammerschmidt, T.G.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Oxidative Stress in Homocystinuria Due to Cystathionine ß-Synthase Deficiency: Findings in Patients and in Animal Models. Cell. Mol. Neurobiol. 2017, 37, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Gallego-Villar, L.; Rivera-Barahona, A.; Oyarzábal, A.; Pérez, B.; Rodríguez-Pombo, P.; Desviat, L.R. Altered Redox Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxid. Med. Cell. Longev. 2018, 2018, 1246069. [Google Scholar] [CrossRef]
- Jacobsen, D.W. Hyperhomocysteinemia and oxidative stress: Time for a reality check? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1182–1184. [Google Scholar] [CrossRef]
- Chan, S.-H.; Hung, C.-H.; Shih, J.-Y.; Chu, P.-M.; Cheng, Y.-H.; Lin, H.-C.; Hsieh, P.-L.; Tsai, K.-L. Exercise intervention attenuates hyperhomocysteinemia-induced aortic endothelial oxidative injury by regulating SIRT1 through mitigating NADPH oxidase/LOX-1 signaling. Redox Biol. 2018, 14, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Sipkens, J.A.; Krijnen, P.A.J.; Hahn, N.E.; Wassink, M.; Meischl, C.; Smith, D.E.C.; Musters, R.J.P.; Stehouwer, C.D.A.; Rauwerda, J.A.; van Hinsbergh, V.W.M.; et al. Homocysteine-induced cardiomyocyte apoptosis and plasma membrane flip-flop are independent of S-adenosylhomocysteine: A crucial role for nuclear p47phox. Mol. Cell. Biochem. 2011, 358, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhang, Z.; Wang, X.; Luo, Z.; Lai, B.; Xiao, L.; Wang, N. Stachydrine protects eNOS uncoupling and ameliorates endothelial dysfunction induced by homocysteine. Mol. Med. 2018, 24, 10. [Google Scholar] [CrossRef] [Green Version]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef]
- Xu, D.; Neville, R.; Finkel, T. Homocysteine accelerates endothelial cell senescence. FEBS Lett. 2000, 470, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Austin, R.C.; Sood, S.K.; Dorward, A.M.; Singh, G.; Shaughnessy, S.G.; Pamidi, S.; Outinen, P.A.; Weitz, J.I. Homocysteine-dependent alterations in mitochondrial gene expression, function and structure. Homocysteine and H2O2 act synergistically to enhance mitochondrial damage. J. Biol. Chem. 1998, 273, 30808–30817. [Google Scholar] [CrossRef] [PubMed]
- Florence, T.M. The production of hydroxyl radical from hydrogen peroxide. J. Inorg. Biochem. 1984, 22, 221–230. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Aldred, S.; Dale, C.; Nakano, E.; Kitas, G.D.; Grant, M.G.; Nugent, D.; Taiwo, F.A.; Li, L.; Powers, H.J. Homocysteine from endothelial cells promotes LDL nitration and scavenger receptor uptake. Free Radic. Biol. Med. 2006, 40, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Nakano, E.; Taiwo, F.A.; Nugent, D.; Griffiths, H.R.; Aldred, S.; Paisi, M.; Kwok, M.; Bhatt, P.; Hill, M.H.E.; Moat, S.; et al. Downstream effects on human low density lipoprotein of homocysteine exported from endothelial cells in an in vitro system. J. Lipid Res. 2005, 46, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Sun, W.; Liu, G.; Zheng, H.; Zhao, J. Comparison of oxidative stress biomarkers in hypertensive patients with or without hyperhomocysteinemia. Clin. Exp. Hypertens. 2018, 40, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Zhang, Y.; Loscalzo, J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J. Biol. Chem. 2005, 280, 15518–15525. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Waly, M.I.; Ali, A.; Al-Nassri, A.; Al-Mukhaini, M.; Valliatte, J.; Al-Farsi, Y. Low nourishment of B-vitamins is associated with hyperhomocysteinemia and oxidative stress in newly diagnosed cardiac patients. Exp. Biol. Med. 2016, 241, 46–51. [Google Scholar] [CrossRef]
- Mohamed, R.; Sharma, I.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.M.; Fulzele, S.; Elmasry, K.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia Alters Retinal Endothelial Cells Barrier Function and Angiogenic Potential via Activation of Oxidative Stress. Sci. Rep. 2017, 7, 11952. [Google Scholar] [CrossRef]
- Esse, R.; Florindo, C.; Imbard, A.; Rocha, M.S.; de Vriese, A.S.; Smulders, Y.M.; Teerlink, T.; Tavares de Almeida, I.; Castro, R.; Blom, H.J. Global protein and histone arginine methylation are affected in a tissue-specific manner in a rat model of diet-induced hyperhomocysteinemia. Biochim. Biophys. Acta 2013, 1832, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Imbard, A.; Florindo, C.; Gupta, S.; Quinlivan, E.P.; Davids, M.; Teerlink, T.; Tavares de Almeida, I.; Kruger, W.D.; Blom, H.J.; et al. Protein arginine hypomethylation in a mouse model of cystathionine β-synthase deficiency. FASEB J. 2014, 28, 2686–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostner, G.M. Lipoprotein Metabolism and Atherogenesis: Implications for Therapy. Cardiology 1991, 78, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, X.; Yang, F.; Gaubatz, J.W.; Ma, L.; Magera, M.J.; Yang, X.; Berger, P.B.; Durante, W.; Pownall, H.J.; et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood 2003, 101, 3901–3907. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Innerarity, T.L.; Mahley, R.W. Role of lysine residues of plasma lipoproteins in high affinity binding to cell surface receptors on human fibroblasts. J. Biol. Chem. 1978, 253, 9053–9062. [Google Scholar] [PubMed]
- Marques, L.R.; Diniz, T.A.; Antunes, B.M.; Rossi, F.E.; Caperuto, E.C.; Lira, F.S.; Gonçalves, D.C. Reverse Cholesterol Transport: Molecular Mechanisms and the Non-medical Approach to Enhance HDL Cholesterol. Front. Physiol. 2018, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Velez-Carrasco, W.; Merkel, M.; Twiss, C.O.; Smith, J.D. Dietary methionine effects on plasma homocysteine and HDL metabolism in mice. J. Nutr. Biochem. 2008, 19, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namekata, K.; Enokido, Y.; Ishii, I.; Nagai, Y.; Harada, T.; Kimura, H. Abnormal Lipid Metabolism in Cystathionine β-Synthase-deficient Mice, an Animal Model for Hyperhomocysteinemia. J. Biol. Chem. 2004, 279, 52961–52969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D.; Tan, H.; Hui, R.; Li, Z.; Jiang, X.; Gaubatz, J.; Yang, F.; Durante, W.; Chan, L.; Schafer, A.I.; et al. Hyperhomocysteinemia Decreases Circulating High-Density Lipoprotein by Inhibiting Apolipoprotein A-I Protein Synthesis and Enhancing HDL Cholesterol Clearance. Circ. Res. 2006, 99, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, H. Homocysteine is a protein amino acid in humans: Implications for homocysteine-linked disease. J. Biol. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein N-homocysteinylation: Implications for atherosclerosis. Biomed. Pharmacother. 2001, 55, 443–447. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine Thiolactone and Protein Homocysteinylation in Human Endothelial Cells: Implications for Atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mercié, P.; Garnier, O.; Lascoste, L.; Renard, M.; Closse, C.; Durrieu, F.; Marit, G.; Boisseau, R.M.; Belloc, F. Homocysteine-thiolactone induces caspase-independent vascular endothelial cell death with apoptotic features. Apoptosis 2000, 5, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najib, S.; Sánchez-Margalet, V. Homocysteine thiolactone inhibits insulin signaling, and glutathione has a protective effect. J. Mol. Endocrinol. 2001, 27, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najib, S.; Sánchez-Margalet, V. Homocysteine thiolactone inhibits insulin-stimulated DNA and protein synthesis: Possible role of mitogen-activated protein kinase (MAPK), glycogen synthase kinase-3 (GSK-3) and p70 S6K phosphorylation. J. Mol. Endocrinol. 2005, 34, 119–126. [Google Scholar] [CrossRef]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical update: Cardiovascular disease in diabetes mellitus. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Meigs, J.B.; Jacques, P.F.; Selhub, J.; Singer, D.E.; Nathan, D.M.; Rifai, N.; D’Agostino, R.B.; Wilson, P.W.F. Fasting Plasma Homocysteine Levels in the Insulin Resistance Syndrome. Diabetes Care 2001, 24, 1403–1410. [Google Scholar] [CrossRef]
- Genoud, V.; Quintana, P.G.; Gionco, S.; Baldessari, A.; Quintana, I. Structural changes of fibrinogen molecule mediated by the N-homocysteinylation reaction. J. Thromb. Thrombolysis 2018, 45, 66–76. [Google Scholar] [CrossRef]
- Sauls, D.L.; Wolberg, A.S.; Hoffman, M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: Implications for the mechanism of thrombosis in hyperhomocysteinemia. J. Thromb. Haemost. 2003, 1, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Boers, G.H.J.; Strauss, K.A. Mutations in cystathionine -synthase or methylenetetrahydrofolate reductase gene increase N-homocysteinylated protein levels in humans. FASEB J. 2008, 22, 4071–4076. [Google Scholar] [CrossRef] [PubMed]
- Szlauer, A.; Mielimonka, A.; Głowacki, R.; Borowczyk, K.; Stachniuk, J.; Undas, A. Protein N-linked homocysteine is associated with recurrence of venous thromboembolism. Thromb. Res. 2015, 136, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Borowczyk, K.; Piechocka, J.; Głowacki, R.; Dhar, I.; Midtun, Ø.; Tell, G.S.; Ueland, P.M.; Nygård, O.; Jakubowski, H. Urinary excretion of homocysteine thiolactone and the risk of acute myocardial infarction in coronary artery disease patients: The WENBIT trial. J. Intern. Med. 2018, 285, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gao, Y.; Zhou, J.; Zhen, Y.; Yang, Y.; Wang, J.; Song, L.; Liu, Y.; Xu, H.; Chen, Z.; et al. Plasma homocysteine thiolactone adducts associated with risk of coronary heart disease. Clin. Chim. Acta 2006, 364, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Borowczyk, K.; Głowacki, R.; Nygård, O.; Jakubowski, H. Paraoxonase 1 Q192R genotype and activity affect homocysteine thiolactone levels in humans. FASEB J. 2018, 32, 6019–6024. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yamaguchi, M.; Motomura, T.; Mizuno, K. Investigation of the relationship between atherosclerosis and paraoxonase or homocysteine thiolactonase activity in patients with type 2 diabetes mellitus using a commercially available assay. Clin. Chim. Acta 2005, 359, 156–162. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef]
- Castro, R.; Rivera, I.; Struys, E.A.; Jansen, E.E.W.; Ravasco, P.; Camilo, M.E.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin. Chem. 2003, 49, 1292–1296. [Google Scholar] [CrossRef]
- Choumenkovitch, S.F.; Selhub, J.; Bagley, P.J.; Maeda, N.; Nadeau, M.R.; Smith, D.E.; Choi, S.-W. In the cystathionine beta-synthase knockout mouse, elevations in total plasma homocysteine increase tissue S-adenosylhomocysteine, but responses of S-adenosylmethionine and DNA methylation are tissue specific. J. Nutr. 2002, 132, 2157–2160. [Google Scholar] [CrossRef]
- Laukkanen, M.O.; Mannermaa, S.; Hiltunen, M.O.; Aittomäki, S.; Airenne, K.; Jänne, J.; Ylä-Herttuala, S. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.O.; Turunen, M.P.; Häkkinen, T.P.; Rutanen, J.; Hedman, M.; Mäkinen, K.; Turunen, A.-M.; Aalto-Setälä, K.; Ylä-Herttuala, S. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc. Med. 2002, 7, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Xie, J.; Zhang, M.; Wang, S. Homocysteine harasses the imprinting expression of IGF2 and H19 by demethylation of differentially methylated region between IGF2/H19 genes. Acta Biochim. Biophys. Sin. 2009, 41, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Ingrosso, D.; Cimmino, A.; Perna, A.F.; Masella, L.; De Santo, N.G.; De Bonis, M.L.; Vacca, M.; D’Esposito, M.; D’Urso, M.; Galletti, P.; et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003, 361, 1693–1699. [Google Scholar] [CrossRef]
- Devlin, A.M.; Bottiglieri, T.; Domann, F.E.; Lentz, S.R. Tissue-specific changes in H19 methylation and expression in mice with hyperhomocysteinemia. J. Biol. Chem. 2005, 280, 25506–25511. [Google Scholar] [CrossRef] [PubMed]
- Han, X.B.; Zhang, H.P.; Cao, C.J.; Wang, Y.H.; Tian, J.; Yang, X.L.; Yang, A.N.; Wang, J.; Jiang, Y.D.; Xu, H. Aberrant DNA methylation of the PDGF gene in homocysteine-mediated VSMC proliferation and its underlying mechanism. Mol. Med. Rep. 2014, 10, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Chen, I.; Yang, F.; Jiang, X.; Jan, M.; Liu, X.; Schafer, A.I.; Durante, W.; Yang, X.; Wang, H. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood 2007, 110, 3648–3655. [Google Scholar] [CrossRef]
- Naushad, S.M.; Prayaga, A.; Digumarti, R.R.; Gottumukkala, S.R.; Kutala, V.K. Bcl-2/adenovirus E1B 19??kDa-interacting protein 3 (BNIP3) expression is epigenetically regulated by one-carbon metabolism in invasive duct cell carcinoma of breast. Mol. Cell. Biochem. 2012, 361, 189–195. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Finer, S.; Voyias, P.D.; McCarthy, C.M.; Vatish, M.; Moore, J.; Smart-Halajko, M.; Bawazeer, N.; Al-Daghri, N.M.; McTernan, P.G.; et al. Vitamin B12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of SREBF1 and LDLR genes. Clin. Epigenet. 2015, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Tekpli, X.; Landvik, N.E.; Anmarkud, K.H.; Skaug, V.; Haugen, A.; Zienolddiny, S. DNA methylation at promoter regions of interleukin 1B, interleukin 6, and interleukin 8 in non-small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 337–345. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, X.; Liu, J.; Xie, X.; Cui, W.; Zhu, Y. Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Hori, H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014, 5, 144. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat. Struct. Mol. Biol. 2015, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Jiang, D.; Wang, Y.; Wang, X. N6-Methyladenosine (m6A) Methylation in mRNA with A Dynamic and Reversible Epigenetic Modification. Mol. Biotechnol. 2016, 58, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; He, C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013, 29, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.; Kapoor, U.; Jantsch, M.F. Understanding RNA modifications: The promises and technological bottlenecks of the “epitranscriptome”. Open Biol. 2017, 7, 170077. [Google Scholar] [CrossRef]
- Glick, J.M.; Ross, S.; Leboy, P.S. S-adenosylhomocysteine inhibition of three purified tRNA methyltransferases from rat liver. Nucleic Acids Res. 1975, 2, 1639–1652. [Google Scholar] [CrossRef]
- Dayal, S.; Brown, K.L.; Weydert, C.J.; Oberley, L.W.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Lentz, S.R. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1996–2002. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. [Google Scholar] [CrossRef]
- Esse, R.; Rocha, M.S.; Barroso, M.; Florindo, C.; Teerlink, T.; Kok, R.M.; Smulders, Y.M.; Rivera, I.; Leandro, P.; Koolwijk, P.; et al. Protein arginine methylation is more prone to inhibition by S-adenosylhomocysteine than DNA methylation in vascular endothelial cells. PLoS ONE 2013, 8, e55483. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Zappia, V.; Galletti, P.; Capasso, G.; De Santo, N.G. Enzymatic methyl esterification of erythrocyte membrane proteins is impaired in chronic renal failure. Evidence for high levels of the natural inhibitor S-adenosylhomocysteine. J. Clin. Investig. 1993, 91, 2497–2503. [Google Scholar] [CrossRef]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.-C.; Perrella, M.A.; Haber, E.; Lee, M.-E. Inhibition of Growth and p21 ras Methylation in Vascular Endothelial Cells by Homocysteine but Not Cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef]
- Garcia, M.M.; Guéant-Rodriguez, R.-M.; Pooya, S.; Brachet, P.; Alberto, J.-M.; Jeannesson, E.; Maskali, F.; Gueguen, N.; Marie, P.-Y.; Lacolley, P.; et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1α by PRMT1 and SIRT1. J. Pathol. 2011, 225, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Wierda, R.J.; Rietveld, I.M.; Van Eggermond, M.C.J.A.; Belien, J.A.M.; Van Zwet, E.W.; Lindeman, J.H.N.; Van Den Elsen, P.J. Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques. Life Sci. 2015, 129, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Greißel, A.; Culmes, M.; Napieralski, R.; Wagner, E.; Gebhard, H.; Schmitt, M.; Zimmermann, A.; Eckstein, H.-H.; Zernecke, A.; Pelisek, J. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb. Haemost. 2015, 114, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.M.; Cook, N.R.; Gaziano, J.M.; Zaharris, E.; MacFadyen, J.; Danielson, E.; Buring, J.E.; Manson, J.A.E. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: A randomized trial. JAMA J. Am. Med. Assoc. 2008, 299, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J.; Meta-Analysis, K.M.A. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carvajal, A.J.; Solà, I.; Lathyris, D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Smulders, Y.M.; Blom, H.J. The homocysteine controversy. J. Inherit. Metab. Dis. 2011, 34, 93–99. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Valli, A.; Carrero, J.J.; Qureshi, A.R.; Garibotto, G.; Bárány, P.; Axelsson, J.; Lindholm, B.; Stenvinkel, P.; Anderstam, B.; Suliman, M.E. Elevated serum levels of S-adenosylhomocysteine, but not homocysteine, are associated with cardiovascular disease in stage 5 chronic kidney disease patients. Clin. Chim. Acta 2008, 395, 106–110. [Google Scholar] [CrossRef]
- Kerins, D.M.; Koury, M.J.; Capdevila, A.; Rana, S.; Wagner, C. Plasma S-adenosylhomocysteine is a more sensitive indicator of cardiovascular disease than plasma homocysteine. Am. J. Clin. Nutr. 2001, 74, 723–729. [Google Scholar] [CrossRef]
- Xiao, Y.; Su, X.; Huang, W.; Zhang, J.; Peng, C.; Huang, H.; Wu, X.; Huang, H.; Xia, M.; Ling, W. Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int. J. Biochem. Cell Biol. 2015, 67, 158–166. [Google Scholar] [CrossRef]
- Dai, Z.; Mentch, S.J.; Gao, X.; Nichenametla, S.N.; Locasale, J.W. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Molecular Mechanism | Species | References | |
|---|---|---|---|
| NADPH oxidase upregulation | M. Musculus, R. norvegicus (cardiomyoblasts), H. Sapiens (cultured ECs) | [52,110,111] | |
| eNOS uncloupling | R. norvegicus, M. Musculus, H. Sapiens | [112,113] | |
| Decreased non-enzymatic antioxidants: | glutathione vitamin B12 vitamin E | H. Sapiens | [107,122] |
| Impaired enzymatic antioxidants: | GPx-1 and -2 Thioredoxin SOD Catalase | M. Musculus, H. Sapiens | [107,108,119,120,123] |
| Species | Observations | References |
|---|---|---|
| H. sapiens | Correlation between circulating AdoHcy, tHcy and global DNA methylation levels | [152,153] |
| H. sapiens | Hcy-induced enhancer CpG hypomethylation at the imprinted H19 locus in human vascular smooth muscle cells; biallelic expression of H19 in patients with renal disease and HHcy | [157,158] |
| H. sapiens | Hcy-induced promoter hypomethylation and mRNA upregulation of the pro-angiogenic gene PDGF (platelet-derived growth factor) | [160] |
| H. sapiens | Hcy-induced DNA hypomethylation of cell cycle progression genes cyclin A and BNIP3 | [161,162] |
| H. sapiens | Hypomethylation of SREBF1 and the LDL receptor gene upon deficiency of vitamin B12 insufficiency | [163] |
| H. sapiens | AdoHcy-induced impaired expression of adhesion molecules and cytokines via inhibition of the EZH2 methyltransferase, leading to decrease in the levels of the repressive histone modification mark H3K27me3 | [33] |
| H. sapiens | Hcy-induced accelerated senescence of endothelial cells via hypomethylation of the telomerase reverse transcriptase gene | [165] |
| H. sapiens | AdoHcy-induced hypomethylation of the selenocysteine-carrying tRNA and altered expression of selenoproteins, including the critical redox regulator GPx-1 | [34] |
| H. sapiens | AdoHcy-induced global protein arginine methylation | [176] |
| H. sapiens | Loss of protein carboxyl methylation in erythrocytes of chronic renal failure patients due to AdoHcy elevation | [177] |
| H. sapiens | Hcy-induced inhibition of carboxyl methylation of p21(ras) in vascular endothelial cells leading to growth inhibition | [178] |
| H. sapiens | Decrease in the levels of the repressive marks H3K9me2, H3K27me2 and H3K27me3 in advanced atherosclerotic plaques | [181,182] |
| R. norvegicus | Decreased global protein arginine methylation in diet-induced HHcy | [124] |
| R. norvegicus | Loss of protein arginine methylation of the PGC-1α transcriptional coactivator in the myocardium upon methyl donor dietary deficiency | [179] |
| M. musculus | Increased expression of H19 in CBS-deficient mice | [159] |
| M. musculus | Decrease global protein arginine methylation in HHcy induced by CBS deficiency | [125] |
| M. musculus | Correlation between the levels of the histone modification mark H3K4me3 in liver and methionine availability in diet | [180] |
| M. musculus, O. cuniculus | Global DNA hypomethylation in atherosclerotic lesions | [155,156] |
| O. cuniculus | Hypomethylation of the antioxidant extracellular SOD gene in atherosclerotic lesions | [155] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. https://doi.org/10.3390/ijms20040867
Esse R, Barroso M, Tavares de Almeida I, Castro R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. International Journal of Molecular Sciences. 2019; 20(4):867. https://doi.org/10.3390/ijms20040867
Chicago/Turabian StyleEsse, Ruben, Madalena Barroso, Isabel Tavares de Almeida, and Rita Castro. 2019. "The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art" International Journal of Molecular Sciences 20, no. 4: 867. https://doi.org/10.3390/ijms20040867
APA StyleEsse, R., Barroso, M., Tavares de Almeida, I., & Castro, R. (2019). The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. International Journal of Molecular Sciences, 20(4), 867. https://doi.org/10.3390/ijms20040867