1. Introduction
It has been recognized since 1974 that ventilation with large tidal volumes and pressure swings can cause alveolar barrier disruption in pre-clinical models [
1]. The clinical implications of ventilator-induced lung injury (VILI) were subsequently confirmed in the landmark ARDSNetwork trial demonstrating that lower tidal volumes and airway pressures in adults with acute respiratory distress syndrome (ARDS) resulted in fewer ventilator days and improved survival [
2], a finding which has since been extended to patients without ARDS [
3,
4]. Despite the appreciation that mechanical ventilation, while life-saving, contributes to and exacerbates lung injury, there remains an incomplete understanding of the mechanism and pathophysiology underlying VILI.
Epithelial injury is a hallmark of VILI, and the loss of alveolar epithelial barrier function leads to alveolar flooding and life-threatening hypoxemia. The human epidermal growth factor receptor-2 (HER2) is expressed by pulmonary epithelial cells, and has recently been implicated in alveolar epithelial dysfunction [
5,
6,
7]. The HER receptor family consists of four plasma membrane-bound tyrosine kinase receptors: HER1 or epidermal growth factor receptor (EGFR), HER2, HER3, and HER4 [
8]. HER2 has no known ligand but can be activated by heterodimerization with any other HER receptors [
9].
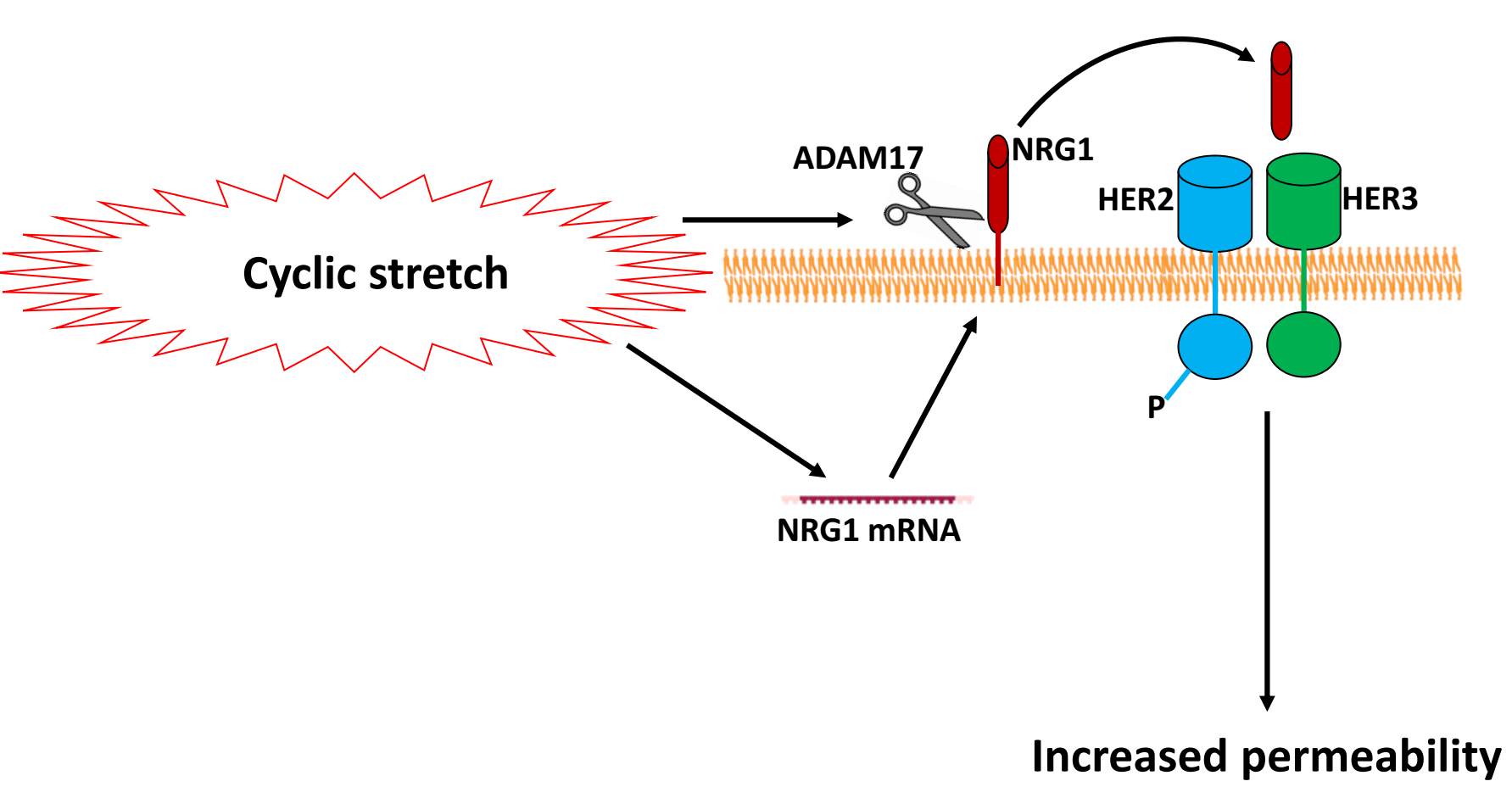
Neuregulin-1 (NRG1) is a HER ligand that is cleaved from pulmonary epithelial cell surfaces by a disintegrin and metalloprotease 17 (ADAM17) [
10]. NRG1-mediated HER2 activation has been implicated in Interleukin-1β (IL-1β)-induced lung injury after bleomycin exposure [
5]. HER3 is a receptor for NRG1, but HER3 has impaired kinase activity and requires heterodimerization with HER2, thereby activating the HER2 tyrosine kinase domain with subsequent HER2 phosphorylation and the initiation of downstream signaling [
11].
Recently, our group performed genome-wide mRNA expression analysis on cyclically-stretched rat alveolar epithelial cells (RAEC) to identify candidate genes involved in stretch-induced increases in permeability [
12]. Given the recent evidence implicating HER ligand-mediated HER2 activation, we queried our database for HER ligands and subsequently investigated the role of HER2 activation in VILI. We hypothesized that HER2 activation contributes to cyclic stretch-induced VILI.
3. Discussion
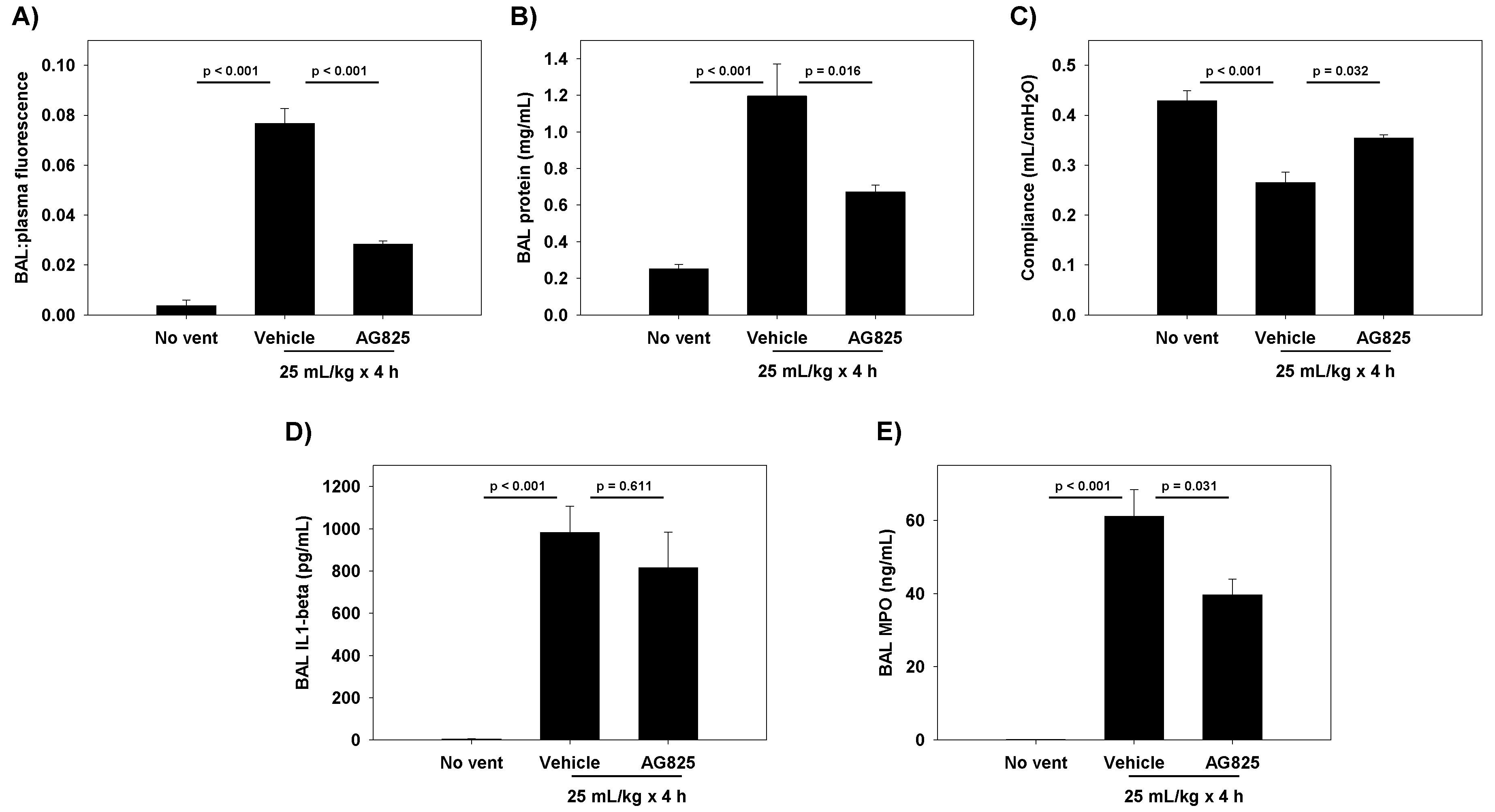
The cyclic stretch of RAEC increased NRG1 expression and release in vitro, with subsequent activation of the HER2 pathway. At the tight junction, ZO-1-bound claudin-7 was slightly reduced, with associated increases in paracellular permeability. The inhibition of NRG1 cleavage and interference with HER3 partially mitigated stretch-induced increases in permeability in vitro, whereas the direct inhibition of HER2 phosphorylation returned the permeability levels to baseline. In rats undergoing damaging ventilation, AG825 mitigated the increased permeability and compliance, suggesting the relevance of HER2 activation in VILI.
Previous studies in bleomycin-injured mice have demonstrated an essential role for NRG1 in linking IL-1β to HER2 activation, and the subsequent downstream disruption of alveolar epithelial barrier function [
5]. The same group has demonstrated elevation in BAL [
5,
6] and plasma [
6] NRG1 in adults with acute respiratory distress syndrome, suggesting that NRG1 can act as a biomarker for lung injury, and implicating NRG1-mediated HER2 activation in diverse inflammatory lung pathologies. We extend these findings to support a potential role for NRG1 and HER2 activation in in vitro and in vivo models of VILI. This has clinical relevance, as it suggests that HER2 remains an active therapeutic target even after the initial inflammatory insult, and further propagates lung injury after the initiation of mechanical ventilation. Our investigations identified four distinct potential targets for intervention: NRG1 expression, NRG1 cleavage by ADAM17, HER3 ligand binding, and HER2 phosphorylation.
Our data did not directly address whether IL-1β mediates NRG1 release, as has been shown with bleomycin-induced lung injury [
5]. The cyclic stretch of epithelial cells in vitro [
14] and injurious ventilation in vivo [
15,
16] are associated with increased IL-1β. However, it is unlikely that NRG1 release is mediated by IL-1β in our model. HER2 activation was noted with as little as 10 min of stretch and was mitigated by TAPI2 or anti-HER3 antibody treatment, suggesting rapid, ADAM17-dependent cleavage of NRG1 after stretch. The cyclic stretch of lung epithelia has only been shown to increase IL-1β after 3 h [
14]. In vivo models of VILI have shown increased IL-1β after 30 to 60 min of ventilation [
15,
16]; however, the source of this IL-1β remains unclear. While both recruited neutrophils [
15] or alveolar macrophages [
17], in addition to stretched lung epithelia, have been implicated in released IL-1β during VILI, in our model, neutrophil infiltration as measured by BAL MPO was reduced with AG825, suggesting other sources of the elevated IL-1β both in animals treated and not treated with AG825. Moreover, while this additional IL-1β potentially contributes to additional NRG1 release during either VILI or any inflammatory lung insult, its impact on the increased NRG1-induced HER2 activation in our in vitro cyclic stretch system is likely negligible. In our in vivo model, the AG825-mediated improvement in permeability could potentially suggest that HER2 activation is downstream of IL-1β. However, this was not explicitly tested.
Our data suggests that NRG1 is released due to increased ADAM17 activation with cyclic stretch, rather than the mechanical disruption of NRG1 binding to cell membranes and subsequent release. Inhibition with TAPI2 reduced HER2 activation and partly mitigated stretch-induced permeability, suggesting a role for increased ADAM17 activity with cyclic stretch. TAPI2 has also been demonstrated to attenuate lung injury induced by IL-1β both in vitro and in vivo [
5], consistent with a central role for ADAM17 in regulating NRG1 cleavage and HER2 activation in diverse etiologies of lung injury. Given the elevated levels of NRG1 mRNA after 6 h of stretch, however, ongoing mechanisms of NRG1 release and paracellular activation are plausible. Furthermore, TAPI2 did not attenuate stretch-induced increases in permeability as well as the direct inhibition of HER2 phosphorylation, suggesting that other pathways leading to HER2 activation, potentially through other HER ligands, may be involved in VILI.
Finigan et al. demonstrated that HER2 activation by NRG1 resulted in phosphorylation of beta-catenin, causing the subsequent dissociation of beta-catenin from E-cadherin and decreased E-cadherin-mediated cell adhesion [
18]. Our results provide another potential pathway leading to increased epithelial permeability via ZO-1-bound claudin-7. However, the magnitude of worsened permeability is larger than the effect on claudin-7. This suggests that there are likely other mechanisms responsible for HER2-mediated increases in permeability.
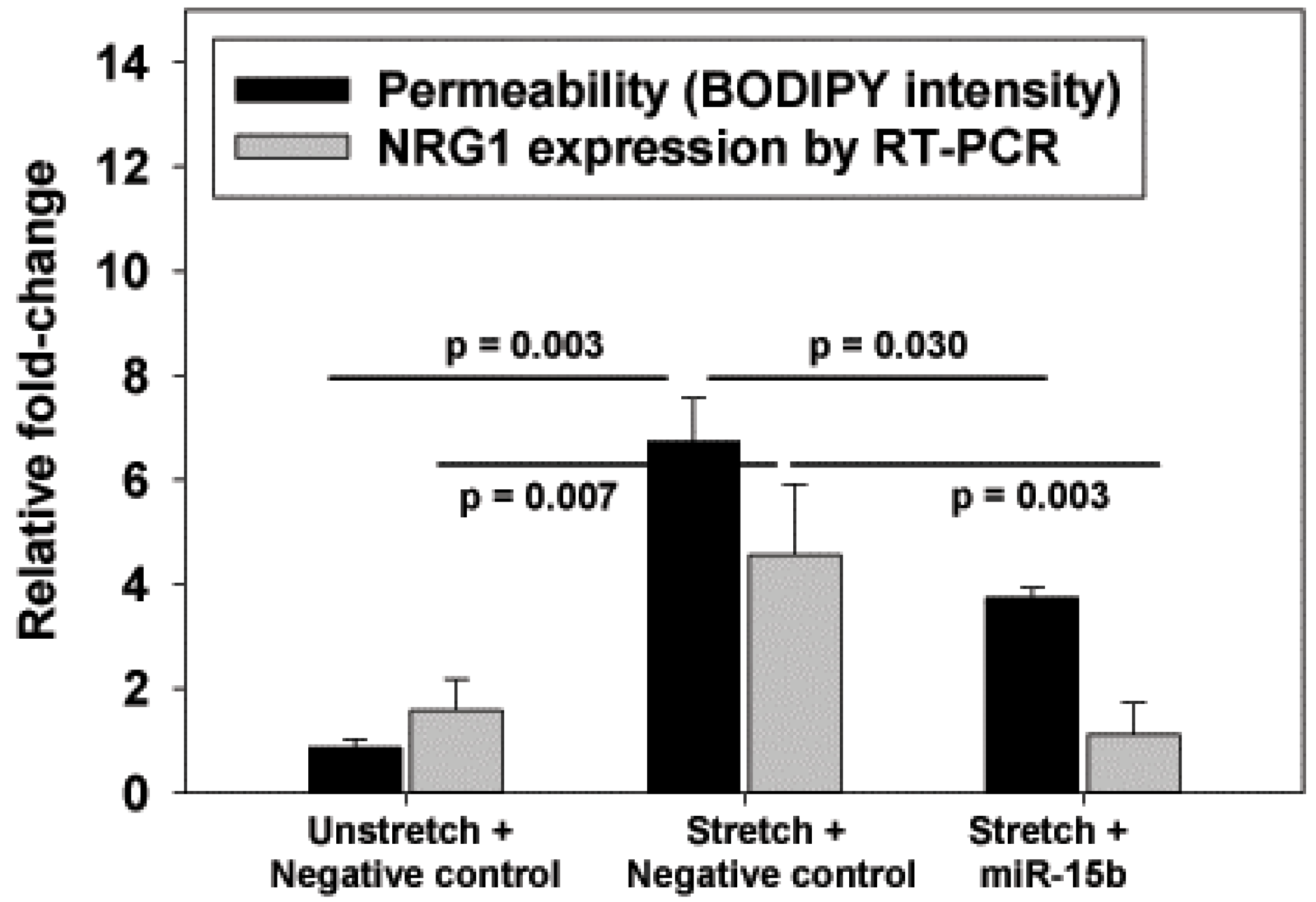
The identification of miR-15b as a possible negative regulator of NRG1 introduces an additional avenue for VILI therapy. As we did not perform a reporter assay to definitively prove that NRG1 mRNA levels were regulated by miR-15b, the improved barrier function with miR-15b may not entirely be associated with reduced NRG1. However, the response to miR-15b and the lower NRG1 mRNA levels were consistent with a role for NRG1 in this pre-clinical model of VILI.
Our study has several limitations. We focused our investigations on NRG1 and the subsequent HER3 transactivation of HER2, as AREG was not elevated. Accordingly, other HER-family receptors, such as EGFR and HER4, were not investigated. Alternative pathways for HER2 activation exist, both involving HER ligands that were not identified in our microarray, as well as non-HER ligands. IL-6, for example, has been shown to activate HER2 by forming a complex with the IL-6 receptor. IL-6, which along with IL-1β is universally elevated after inflammatory stimuli, may also contribute to HER2 activation in vivo. Likely, multiple inflammatory stimuli converge on HER2 activation, which we demonstrate plays a central role in disrupted alveolar barrier function in experimental lung injury. In vivo inhibition of HER2 activation with AG825 in our model of injurious ventilation improved permeability, suggesting that HER2 activation was sufficiently upstream to warrant targeting in VILI and other etiologies of lung injuries. However, we did not assess whether AG825 resulted in lower levels of pHER2 in vivo, which limited our conclusions. Additionally, we could not successfully perform ZO-1 IP in cells treated with anti-HER3 antibodies, preventing an assessment of direct HER3 inhibition on claudin levels. Finally, we did not collect blood gases or histology, precluding a full assessment of the damage caused by our VILI model, and the complete nature of the protection offered by AG825.
4. Methods
4.1. Rat Alveolar Epithelial Cell Isolation
In a protocol approved by the Institutional Animal Care and Use Committee (IACUC protocol 804154, last approved 27 December 2017) of the University of Pennsylvania, alveolar type II cells were isolated from male Sprague-Dawley rats based on a method reported by Dobbs et al. [
19] with a slight modification [
20]. Briefly, rat lungs were isolated while continuously ventilated and perfused via the pulmonary artery to remove blood immediately following sacrifice. Lungs underwent alveolar lavage to remove debris and subsequently underwent enzymatic digestion followed by mincing. The resultant solution was depleted of immune cells by negative selection following incubation in IgG-coated plates, resulting in type II cells. Type II cells were seeded onto fibronectin coated (10 μg/cm
2) flexible silastic membranes (Specialty Manufacturing, Saginaw, MI, USA), and mounted in custom designed wells at a density of 10
6 cells/cm
2. Cells were cultured for 5 days with MEM supplemented with 10% fetal bovine serum (FBS), until they attained alveolar type I cell characteristics [
12,
13,
21].
4.2. Cyclic Stretch
Type I-like RAEC were serum-deprived with 20 mM Hepes supplemented with DMEM (CO2 free buffering system) for 2 h, and subjected to biaxial cyclic stretch (∆SA 25% or ΔSA 37%) at 37 °C at a frequency of 0.25 Hz (15 cycles/min). Cells were stretched for 10 min (phosphorylation experiments), 1 h (HER ligand secretion experiments), or 6 h (gene expression and miRNA transfection experiments). In some experiments, cells were treated for 2 h prior to and during stretch with either the ADAM17 inhibitor TAPI2 (50 µM; Calbiochem, Burlington, MA, USA), competitive anti-HER3 antibody to prevent ligand binding (10 µg antibody/mL DMEM; Abcam, Cambridge, MA, USA), the HER2 tyrosine kinase inhibitor AG825 (50 µM; Calbiochem), or vehicle control (DMSO).
4.3. RT-PCR
Total RNA was extracted from the cells (Qiagen miRNAeasy mini kit cat# 217004, Qiagen Inc, Valencia, CA, USA) per manufacturer’s instructions. The quantity and quality of the RNA samples was measured (Agilent Bioanalyzer and Nanodrop spectrophotometer, Santa Clara, CA, USA). First strand cDNA synthesis occurred in 20 µL reactions with sequence specific Taqman primers, per manufacturer’s protocol. PCR was performed using Taqman universal master mix. Rat 4.5s RNA was used as an endogenous control for miRNA RT-PCR, and GAPDH used for mRNA. Values were normalized to endogenous controls using the ΔCt method, and subsequently to the unstretched control group.
4.4. Western Blotting of Secreted HER Ligands
After 1 h of cyclic stretch at ΔSA 37% (or unstretched control condition), RAEC media were collected, protein concentrated, and quantified by Bradford assay. Equal amounts (60 µg) of the protein was run on SDS-polyacrylamide (4–12%) gels, transferred onto polyvinylidene fluoride membranes (PVDF), and non-specific binding was blocked in TBS containing 5% non-fat powdered milk and 0.1% Tween-20 at room temperature. Membranes were probed for AREG or NRG1 (both antibodies from Santa Cruz Biotechnology, Dallas, TX, USA). Specific activity was calculated through densitometric analysis (Kodak, Rochester, NY, USA). Band intensities were normalized to unstretched control cells.
4.5. Co-Immunoprecipitation and Western Blotting
After 10 min of cyclic stretch at ΔSA 37% (or unstretched control condition), RAEC were washed with PBS and scraped from silastic membranes. To measure active phosphorylated HER2 (pHER2), we used the Pierce Classic Immunoprecipitation (IP) Kit to enrich for pHER2. Given that NRG1 binds HER3 and induces heterodimerization with HER2, we performed IP (50 µg bait antibody) using (separately) either anti-HER3 or anti-HER2 (both antibodies from Cell Signaling Technologies, Danvers, MA, USA). After elution of immune complex, 60 µg of the protein lysate was run on SDS-polyacrylamide (4–12%) gels, transferred onto PVDF, and non-specific binding blocked in TBS containing 5% non-fat powdered milk and 0.1% Tween-20 at room temperature. Membranes were probed for pHER2 (phospho-Tyr1248, Cell Signaling Technologies).
A similar method was used to measure tight junction proteins binding to ZO-1. IP was performed using anti-ZO-1 (Cell Signaling Technologies), and Western blotting performed as above probing for occludin (Zymed/Invitrogen, Waltham, MA, USA), claudin-4, claudin-7, or claudin-18 (all claudin antibodies from Zymed/Invitrogen). Band intensities for probed proteins were normalized to unstretched controls.
4.6. MicroRNA Transfection
Using above described methods, we isolated RAEC and cultured it for 3 days in antibiotic free MEM plus 10% FBS. On day 4 of culture, we transfected a custom-designed miR-15b mimic, or a scrambled negative control (Exiquon, Vedbaek, Denmark). These constructs use a locked nucleic acid (LNA) technology, and were conjugated to a Texas Red dye for visual confirmation of transfection efficiency. RAEC were transfected with miR-15b or negative control using Lipofectamine 2000 (Invitrogen) reagent diluted in Opti-MEM for a final miRNA concentration of 80 nM, per manufacturer’s protocol. Optimum transfection concentration was determined by titration of different concentrations of inhibitors (50 to 100 nM) and evaluating monolayer fluorescence after 24 h, using >90% cellular fluorescence as a threshold for efficient transfection. On day 5, RAEC were serum-deprived with 20 mM Hepes supplemented with DMEM for 2 h, subjected to biaxial cyclic stretch (∆SA 25%) at 37 °C for 6 hat a frequency of 0.25 Hz (15 cycles/min), or served as unstretched controls.
4.7. Cell Permeability Assays
To test RAEC permeability, we used ouabain, a small uncharged molecule, with high affinity for basolateral Na
+/K
+-ATPase, which when conjugated to the nonpolar fluorophore BODIPY, served as a tracer. Our group has validated this method before [
13,
22,
23], and demonstrated that BODIPY-ouabain binding represents loss of tight junction integrity and increased paracellular permeability, and that increased fluorescent signal is not a result of cell death or intracellular uptake or transport. BODIPY-ouabain (Invitrogen) diluted in dimethyl sulfoxide was added to cells at a final concentration of 2 µM, and incubated for 1 h. At the end of the hour, all cells were washed with DMEM to remove excess BODIPY-ouabain from the apical surface, and were visualized under a fluorescent microscope using a green emission filter. Presence of fluorescence was evidence of high affinity ouabain binding to the basolateral surface, and used as a measure of paracellular permeability. Fluorescence was measured on 4 separate fields per well, and 3 wells were measured per treatment group per animal.
4.8. In Vivo Model of Ventilator-Induced Lung Injury
This protocol was approved by the University of Pennsylvania IACUC. Male Sprague-Dawley rats (aged 8–10 weeks) were randomized to receive either vehicle (DMSO) or AG825 (1.67 mg/kg/day) intraperitoneally (0.5 mL daily) for 3 total doses (total 5 mg/kg). On day 3, rats were anesthetized with pentobarbital (50 mg/kg intraperitoneal), underwent tracheostomy with a stiff 14G catheter, and received a subcutaneous 10 mL saline bolus. FITC-labeled albumin (Sigma Aldrich) was intravenously injected (0.5 mL of 15 mg/mL solution diluted in PBS) prior to ventilation. Rats underwent injurious ventilation for 4 h (VT 25 mL/kg, end-expiratory pressure 0 cmH2O, rate 27 breaths/minute, FIO2 0.21) using a small-animal ventilator (Harvard Apparatus, Holliston, MA, USA). Non-ventilated, vehicle-treated rats served as a comparison group. Respiratory system static compliance was assessed by attaching the tracheal cannula to a pressure manometer/capnograph (Harvard Apparatus), and plateau pressure recorded at the end of 2 h of ventilation during an inspiratory hold during volume control ventilation, with tidal volume recorded within the ventilator. Non-ventilated control rats underwent tracheal cannulation and attachment to a pressure manometer and ventilator for measurement of compliance similar to ventilated rats.
At the end of the experiment, blood was collected via ventricular puncture into citrated tubes, and rats sacrificed by exsanguination. Citrated blood was centrifuged (2000 g, 20 min, 20 °C) to generate platelet-poor plasma. Rats underwent BAL using 24 mL of PBS (Ca2+ and Mg2+ added) in 3 separate aliquots of 8 mL each. Each aliquot was centrifuged (300 g, 10 min, 10 °C), and the cell-free supernatant recovered. Fluorescence was measured in both the BAL supernatant and the plasma, and permeability was assessed by the ratio of BAL: blood fluorescence. BAL total protein levels were measured by the DC Protein Assay (BioRad, Hercules, CA, USA). BAL levels of IL-1β were measured using Rat IL-1β ELISA (RayBiotech Inc., Norcross, GA, USA). BAL neutrophil activity was quantified using a myeloperoxidase (MPO) ELISA (R & D Systems, Minneapolis, MN, USA).
4.9. Statistical Analysis
Values are expressed as means ± SEM. Parametric statistics, including unpaired t-tests or ANOVA were used as appropriate. ANOVA was followed by post-hoc Tukey test. p < 0.05 was considered statistically significant. To standardize reporting of different measurements in RAEC, (RNA by RT-PCR, protein intensity by Western, fluorescence intensity by imaging), values are normalized to unstretched control cells.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}