Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cytotoxicity Analyses, Growth Assessments, Morphology, and Surface Marker Expression of MV4-11-R
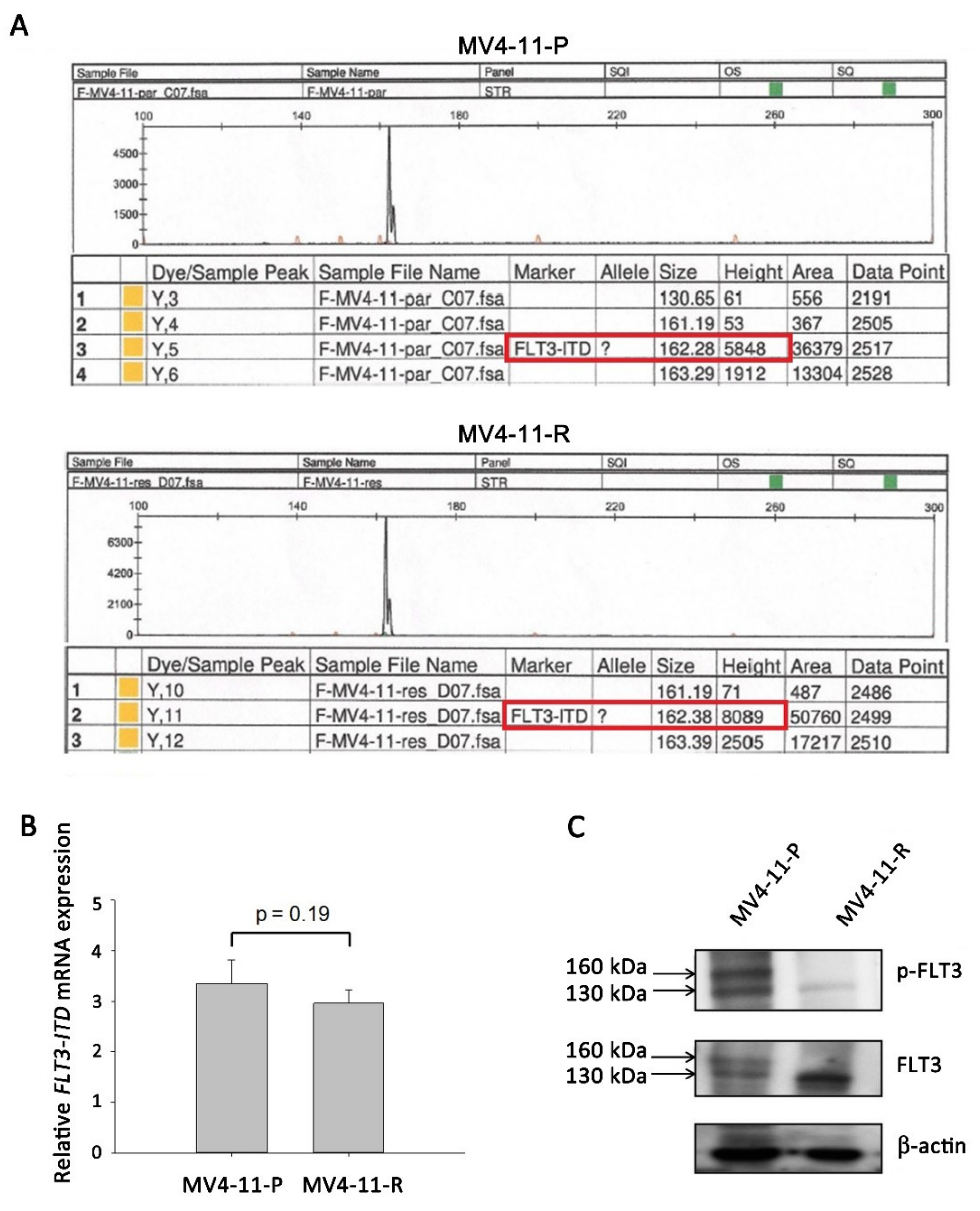
2.2. FLT3-ITD Mutation and Activation Status in MV4-11-R
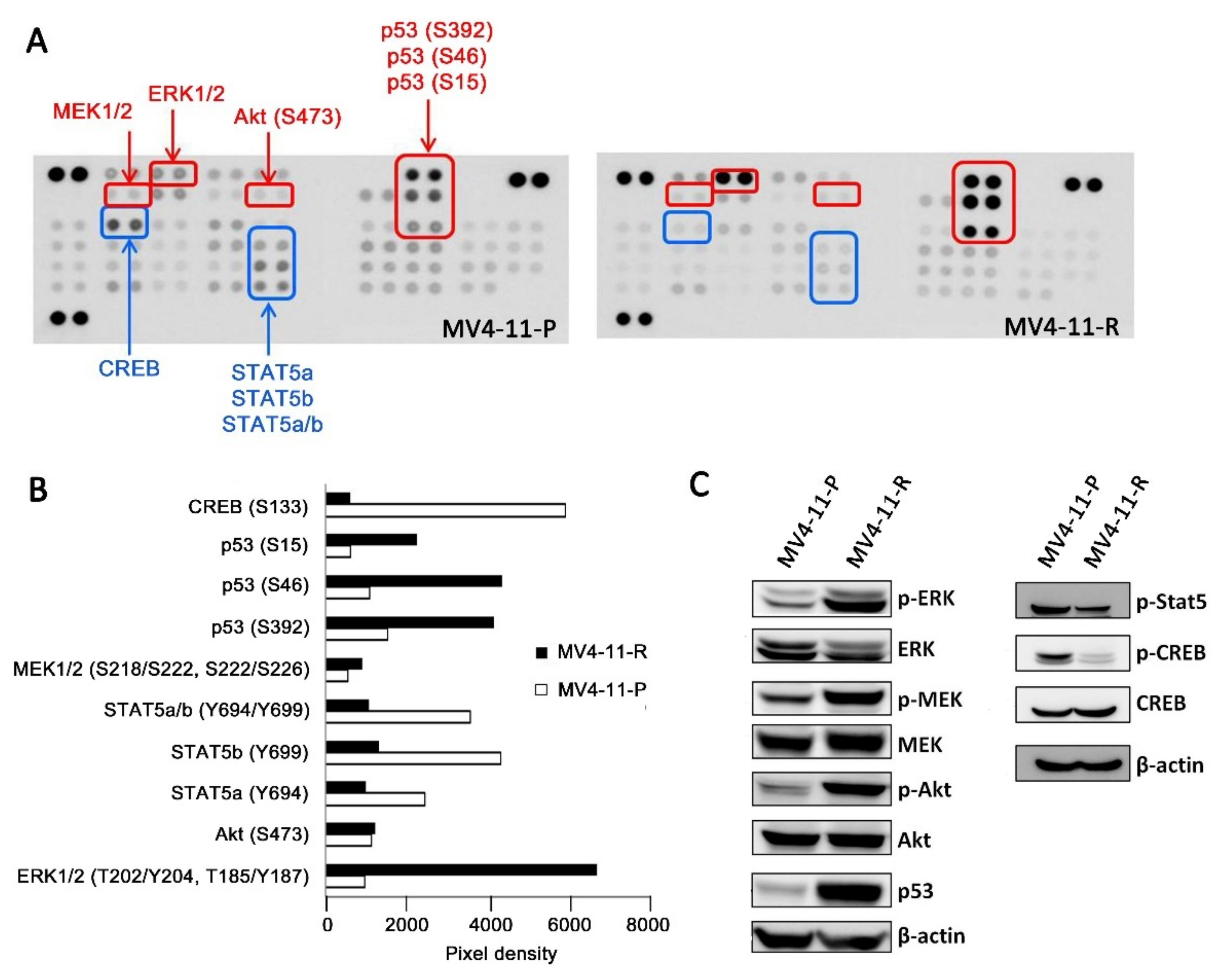
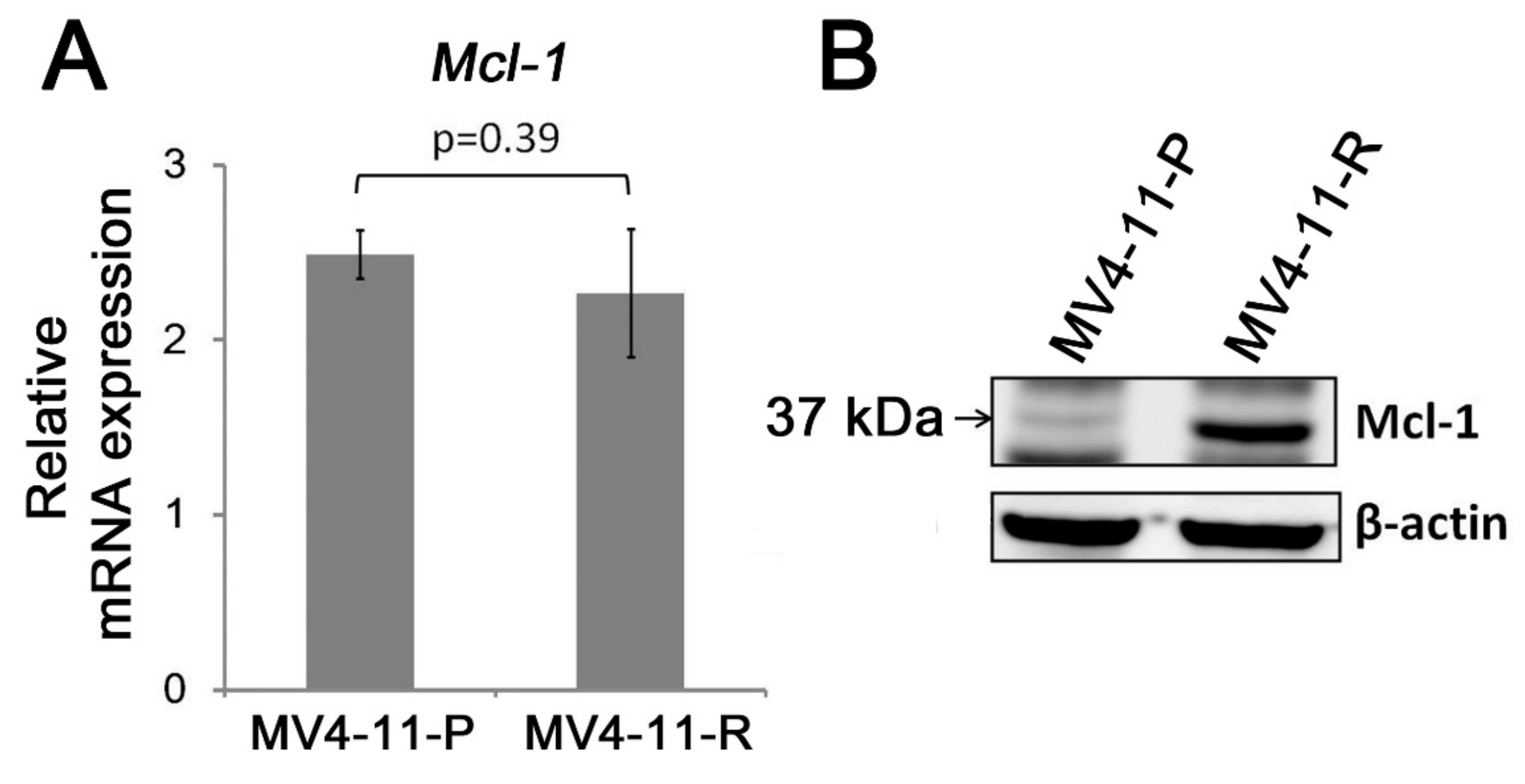
2.3. Apoptosis-Related Proteins in MV4-11-R
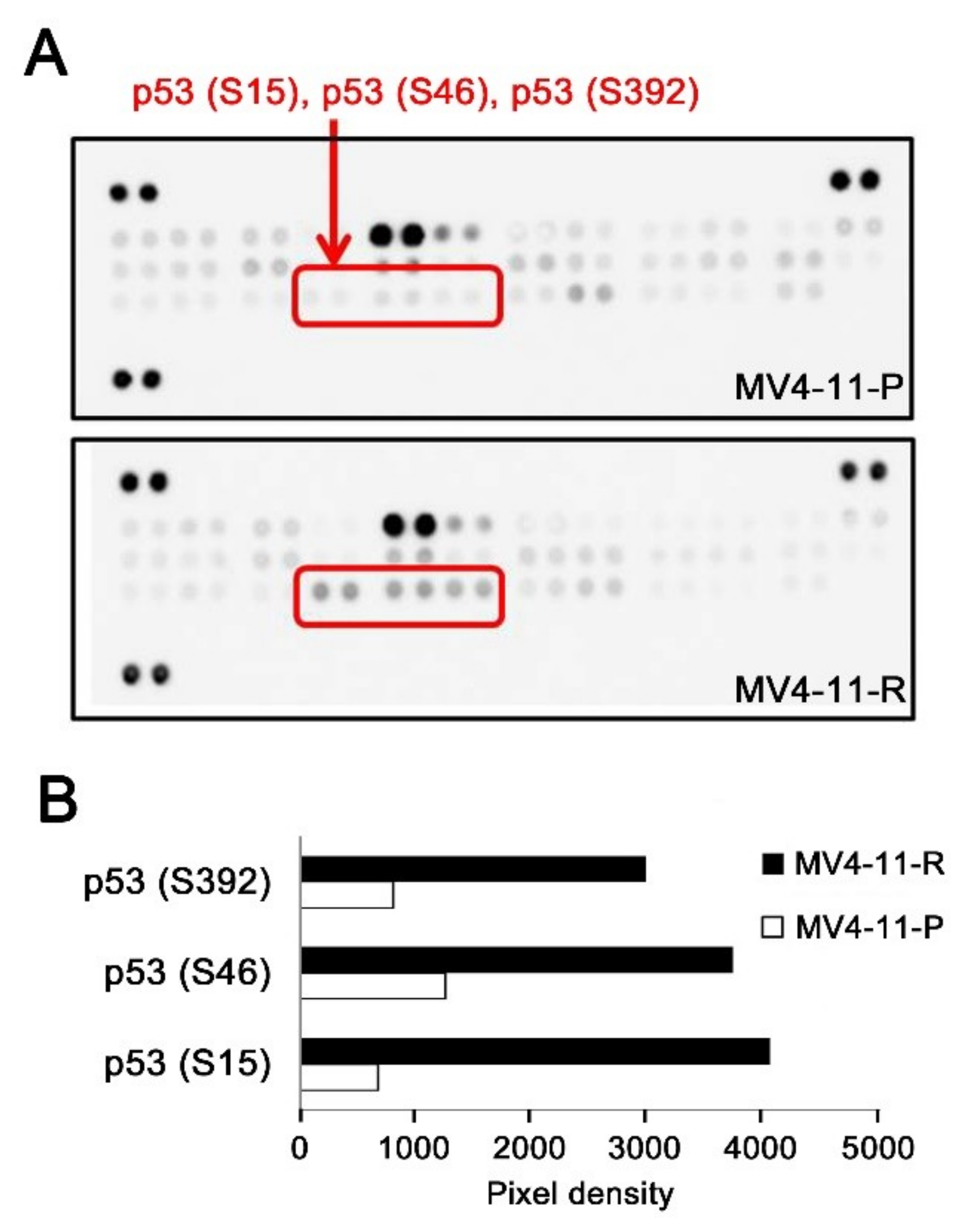
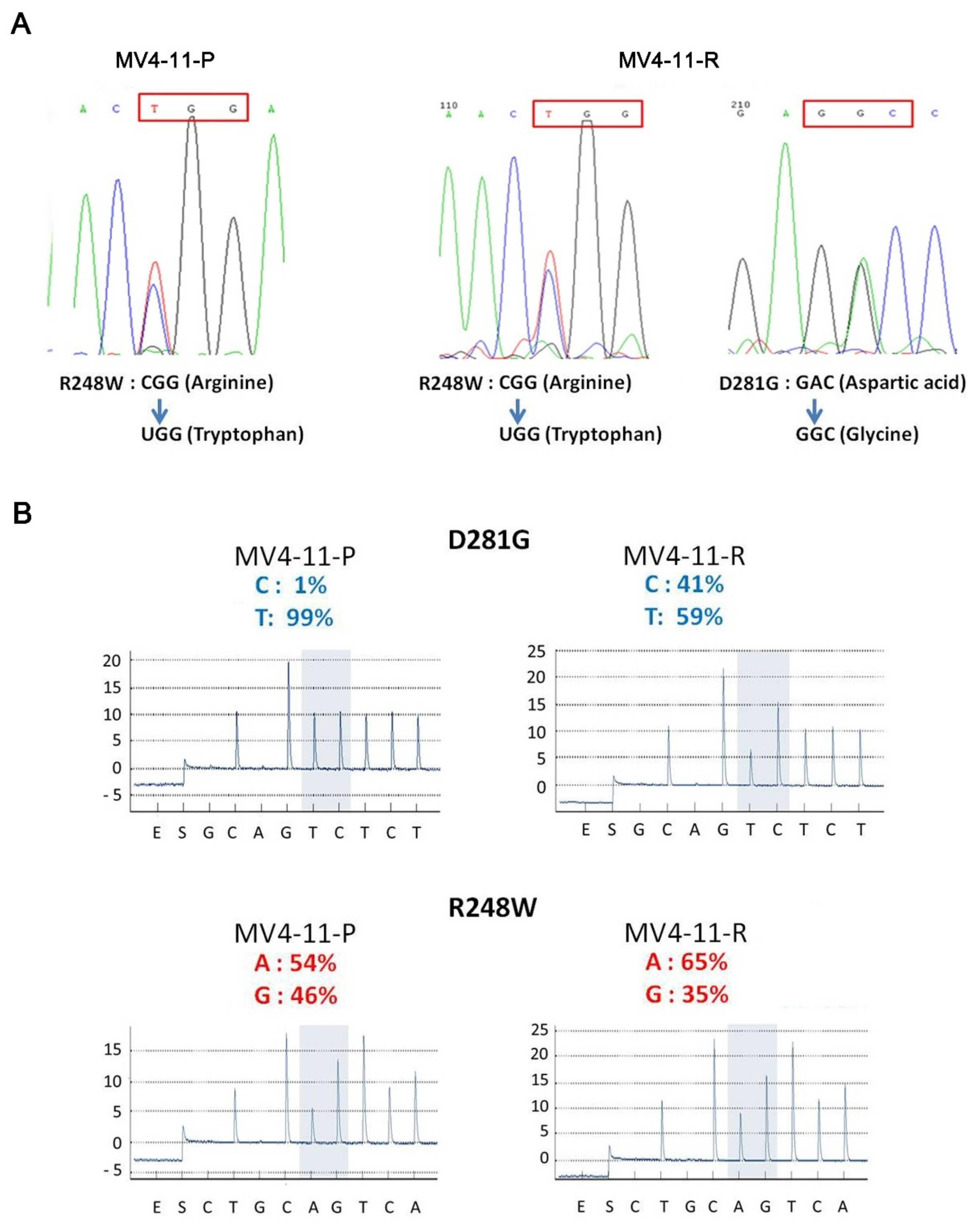
2.4. An Additional TP53 Mutation Emerged in MV4-11-R
2.5. Examination of the Cytarabine Metabolic Pathway and Multidrug Resistance Genes in MV4-11-R
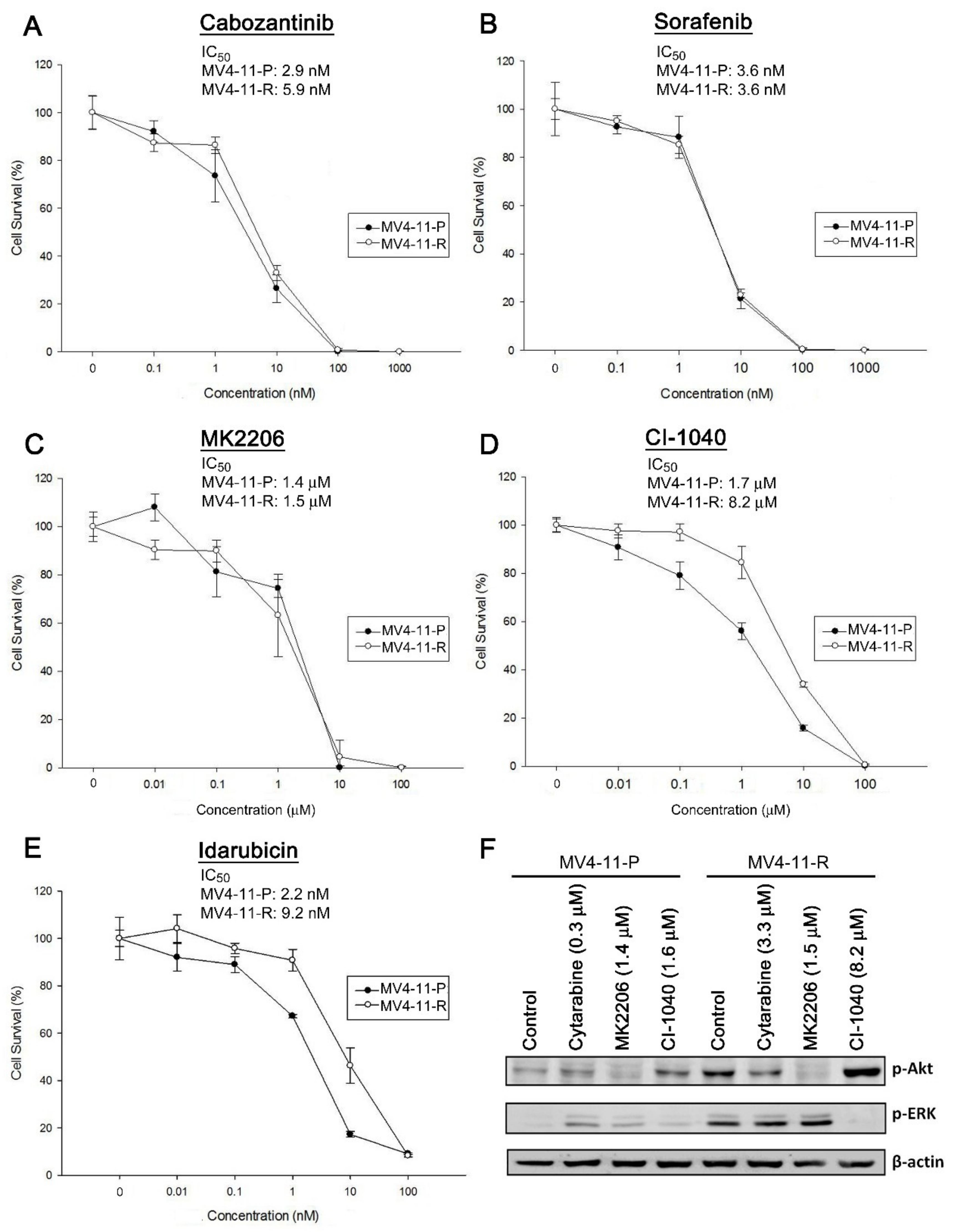
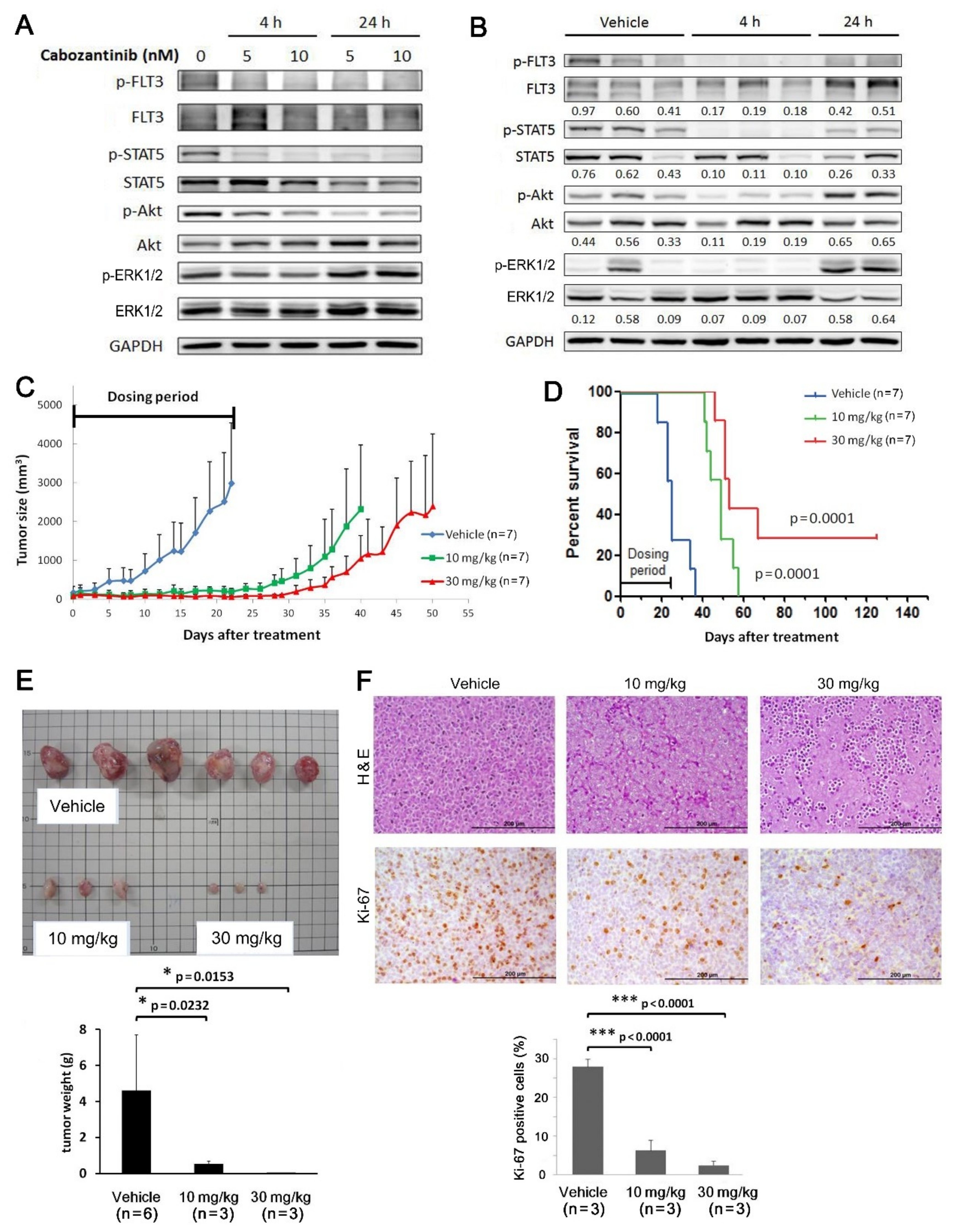
2.6. Cabozantinib Effectively Inhibits Tumorigenic Features of MV4-11-P and MV4-11-R Both In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Establishment of the Cytarabine-Resistant Cell Line
4.2. Cytotoxicity Curves
4.3. Cytogenetics
4.4. RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR (qPCR)
4.5. DNA Extraction, GeneScan Analysis, and DNA Sequencing
4.6. Pyrosequencing
4.7. Western Blot Analysis
4.8. Analysis of Kinase Phosphorylation and Apoptosis-Related Protein Profiles
4.9. Mouse Xenograft Experiments
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perl, A.E. The role of targeted therapy in the management of patients with AML. Blood Adv. 2017, 1, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Cros, E.; Jordheim, L.; Dumontet, C.; Galmarini, C.M. Problems related to resistance to cytarabine in acute myeloid leukemia. Leuk. Lymphoma 2004, 45, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Kim, S.H.; Kweon, S.H.; Lee, T.H.; Kim, H.J.; Kim, H.J.; Kim, S. Defective expression of deoxycytidine kinase in cytarabine-resistant acute myeloid leukemia cells. Int. J. Oncol. 2009, 34, 1165–1171. [Google Scholar] [PubMed]
- Mansson, E.; Paul, A.; Lofgren, C.; Ullberg, K.; Paul, C.; Eriksson, S.; Albertioni, F. Cross-resistance to cytosine arabinoside in a multidrug-resistant human promyelocytic cell line selected for resistance to doxorubicin: Implications for combination chemotherapy. Br. J. Haematol. 2001, 114, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Legrand, O.; Simonin, G.; Beauchamp-Nicoud, A.; Zittoun, R.; Marie, J.P. Simultaneous activity of MRP1 and Pgp is correlated with in vitro resistance to daunorubicin and with in vivo resistance in adult acute myeloid leukemia. Blood 1999, 94, 1046–1056. [Google Scholar] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Van Linden, A.A.; Baturin, D.; Ford, J.B.; Fosmire, S.P.; Gardner, L.; Korch, C.; Reigan, P.; Porter, C.C. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol. Cancer Ther. 2013, 12, 2675–2684. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.; Hiura, T.; Shouji, A.; Osanai, Y.; Ujibe, M.; Ishikawa, M. Resistance to Ara-C up-regulates the activation of NF-kappaB, telomerase activity and Fas expression in NALM-6 cells. Biol. Pharm. Bull. 2007, 30, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Kogan, S.C.; Dickins, R.A.; Lowe, S.W.; Largaespada, D.A. Trp53 loss during in vitro selection contributes to acquired Ara-C resistance in acute myeloid leukemia. Exp. Hematol. 2006, 34, 631–641. [Google Scholar]
- Rassidakis, G.Z.; Herold, N.; Myrberg, I.H.; Tsesmetzis, N.; Rudd, S.G.; Henter, J.I.; Schaller, T.; Ng, S.B.; Chng, W.J.; Yan, B.; et al. Low-level expression of SAMHD1 in acute myeloid leukemia (AML) blasts correlates with improved outcome upon consolidation chemotherapy with high-dose cytarabine-based regimens. Blood Cancer J. 2018, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.G.; Sanjiv, K.; Kutzner, J.; Myrberg, I.H.; Paulin, C.B.J.; Olsen, T.K.; Helleday, T.; Henter, J.I.; Schaller, T. With me or against me: Tumor suppressor and drug resistance activities of SAMHD1. Exp. Hematol. 2017, 52, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Annesley, C.E.; Brown, P. The Biology and Targeting of FLT3 in Pediatric Leukemia. Front. Oncol. 2014, 4, 263. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.C.; Druley, T.E.; Erez, A.; Kulper, R.P.; Onel, K.; Schiffman, J.D.; Wolfe Schneider, K.; Scollon, S.R.; Scott, H.S.; Strong, L.C.; et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin. Cancer Res. 2017, 23, e14–e22. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Matsushita, H.; Asai, S.; Tsukamoto, H.; Ono, R.; Nosaka, T.; Yahata, T.; Takahashi, S.; Miyachi, H. FLT3-ITD induces ara-C resistance in myeloid leukemic cells through the repression of the ENT1 expression. Biochem. Biophys. Res. Commun. 2009, 390, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Reinhardt, J.; Zaborski, M.; Drexler, H.G. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 2003, 17, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Tchelebi, L.; Ashamalla, H.; Graves, P.R. Mutant p53 and the response to chemotherapy and radiation. Subcell. Biochem. 2014, 85, 133–159. [Google Scholar] [PubMed]
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57, 4285–4300. [Google Scholar] [PubMed]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- CancerDR: Cancer Drug Resistance Database. Available online: http://crdd.osdd.net/raghava/cancerdr/index.html (accessed on 22 December 2018).
- Genomics of Drug Sensitivity in Cancer. Available online: https://www.cancerrxgene.org/ (accessed on 14 January 2019).
- Lu, J.W.; Wang, A.N.; Liao, H.A.; Chen, C.Y.; Hou, H.A.; Hu, C.Y.; Tien, H.F.; Ou, D.L.; Lin, L.I. Cabozantnib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A. Clinical pharmacokinetics of nucleoside analogues: Focus on haematological malignancies. Clin. Pharmacokinet. 2000, 39, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Ozkaynak, M.F.; Avramis, V.I.; Carcich, S.; Ortega, J.A. Pharmacology of cytarabine given as a continuous infusion followed by mitoxantrone with and without amsacrine/etoposide as reinduction chemotherapy for relapsed or refractory pediatric acute myeloid leukemia. Med. Pediatric Oncol. 1998, 31, 475–482. [Google Scholar] [CrossRef]
- Van Prooijen, H.C.; Dekker, A.W.; Punt, K. The use of intermediate dose cytosine arabinoside (ID Ara-C) in the treatment of acute non-lymphocytic leukaemia in relapse. Br. J. Haematol. 1984, 57, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; den Boer, M.L.; Meijerink, J.P.; Ebus, M.E.; Peters, G.J.; Noordhuis, P.; Janka-Schaub, G.E.; Armstrong, S.A.; Korsmeyer, S.J.; Pieters, R. Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia. Blood 2003, 101, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Ramakers-van Woerden, N.L.; Beverloo, H.B.; Veerman, A.J.; Camitta, B.M.; Loonen, A.H.; van Wering, E.R.; Slater, R.M.; Harbott, J.; den Boer, M.L.; Ludwig, W.D.; et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia 2004, 18, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.E.; Bohmer, A.; Markova, B.; Choudhary, C.; Serve, H.; Bohmer, F.D. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol. Cell. Biol. 2005, 25, 3690–3703. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Fukushima, H.; Shaik, S.; Liu, P.; Lau, A.W.; Wei, W. Mcl-1 ubiquitination and destruction. Oncotarget 2011, 2, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Lin, Y.M.; Lai, Y.L.; Chen, C.Y.; Hu, C.Y.; Tien, H.F.; Lin, L.I. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med. Oncol. 2015, 32, 206. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Breitenbuecher, F.; Heidel, F.; Hoffarth, S.; Markova, B.; Schuler, M.; Fischer, T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.R.; Wei, J.; Konopleva, M.; Reed, J.C.; Pellecchia, M.; Andreeff, M.; Ruvolo, P.P. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 2015, 126, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Grant, S. Mcl-1 as a Therapeutic Target in Acute Myelogenous Leukemia (AML). Leuk. Res. Rep. 2013, 2, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; Liu, C.W.; Tang, J.L.; et al. TP53 mutations in de novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 1998, 18, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Jackson, C.T.; Subler, M.A.; Martin, D.W. Modulation of cellular and viral promoters by mutant human p53 proteins found in tumor cells. J. Virol. 1992, 66, 6164–6170. [Google Scholar] [PubMed]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 1996, 16, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Weisz, L.; Zalcenstein, A.; Stambolsky, P.; Cohen, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res. 2004, 64, 8318–8327. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Santamaria, A.B.; Bolshakov, S.V.; Ananthaswamy, H.N. Mutant p53 is constitutively phosphorylated at Serine 15 in UV-induced mouse skin tumors: Involvement of ERK1/2 MAP kinase. Oncogene 2003, 22, 5958–5966. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, L.F.; Wang, Y.; Correa, R.G.; Cho, J.Y.; Libermann, T.A. Blockage of NF-kappaB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Nanbakhsh, A.; Pochon, C.; Mallavialle, A.; Amsellem, S.; Bourhis, J.H.; Chouaib, S. c-Myc regulates expression of NKG2D ligands ULBP1/2/3 in AML and modulates their susceptibility to NK-mediated lysis. Blood 2014, 123, 3585–3595. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Nayak, R.; Grant, S. Isolation and characterization of a deoxycytidine kinase-deficient human promyelocytic leukemic cell line highly resistant to 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1984, 44, 5029–5037. [Google Scholar] [PubMed]
- Rathe, S.K.; Largaespada, D.A. Deoxycytidine kinase is downregulated in Ara-C-resistant acute myeloid leukemia murine cell lines. Leukemia 2010, 24, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Negoro, E.; Yamauchi, T.; Urasaki, Y.; Nishi, R.; Hori, H.; Ueda, T. Characterization of cytarabine-resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int. J. Oncol. 2011, 38, 911–919. [Google Scholar] [PubMed]
- Herold, N.; Rudd, S.G.; Ljungblad, L.; Sanjiv, K.; Myrberg, I.H.; Paulin, C.B.; Heshmati, Y.; Hagenkort, A.; Kutzner, J.; Page, B.D.; et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat. Med. 2017, 23, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Malani, D.; Murumagi, A.; Yadav, B.; Kontro, M.; Eldfors, S.; Kumar, A.; Karjalainen, R.; Majumder, M.M.; Ojamies, P.; Pemovska, T.; et al. Enhanced sensitivity to glucocorticoids in cytarabine-resistant AML. Leukemia 2017, 31, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Veuger, M.J.; Honders, M.W.; Spoelder, H.E.; Willemze, R.; Barge, R.M. Inactivation of deoxycytidine kinase and overexpression of P-glycoprotein in AraC and daunorubicin double resistant leukemic cell lines. Leukemia Res. 2003, 27, 445–453. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.; Dahlberg, N.; Tidefelt, U.; Paul, C.; Andersson, G. Characterization of an anthracycline-resistant human promyelocyte leukemia (HL-60) cell line with an elevated MDR-1 gene expression. Biochem. Pharmacol. 1995, 49, 755–762. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Megias-Vericat, J.E.; Martinez-Cuadron, D.; Herrero, M.J.; Alino, S.F.; Poveda, J.L.; Sanz, M.A.; Montesinos, P. Pharmacogenetics of metabolic genes of anthracyclines in acute myeloid leukemia. Curr. Drug Metab. 2018, 19, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Tien, H.F.; Wang, C.H.; Lin, M.T.; Lee, F.Y.; Liu, M.C.; Chuang, S.M.; Chen, Y.C.; Shen, M.C.; Lin, K.H.; Lin, D.T. Correlation of cytogenetic results with immunophenotype, genotype, clinical features, and ras mutation in acute myeloid leukemia. A study of 235 Chinese patients in Taiwan. Cancer Genet. Cytogenet. 1995, 84, 60–68. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, Y.-C.; Hu, C.-Y.; Liu, Z.-H.; Tien, H.-F.; Ou, D.-L.; Chien, H.-F.; Lin, L.-I. Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib. Int. J. Mol. Sci. 2019, 20, 1230. https://doi.org/10.3390/ijms20051230
Ko Y-C, Hu C-Y, Liu Z-H, Tien H-F, Ou D-L, Chien H-F, Lin L-I. Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib. International Journal of Molecular Sciences. 2019; 20(5):1230. https://doi.org/10.3390/ijms20051230
Chicago/Turabian StyleKo, Ya-Chen, Chung-Yi Hu, Zheng-Hau Liu, Hwei-Fang Tien, Da-Liang Ou, Hsiung-Fei Chien, and Liang-In Lin. 2019. "Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib" International Journal of Molecular Sciences 20, no. 5: 1230. https://doi.org/10.3390/ijms20051230
APA StyleKo, Y. -C., Hu, C. -Y., Liu, Z. -H., Tien, H. -F., Ou, D. -L., Chien, H. -F., & Lin, L. -I. (2019). Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib. International Journal of Molecular Sciences, 20(5), 1230. https://doi.org/10.3390/ijms20051230