Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module

 and
and 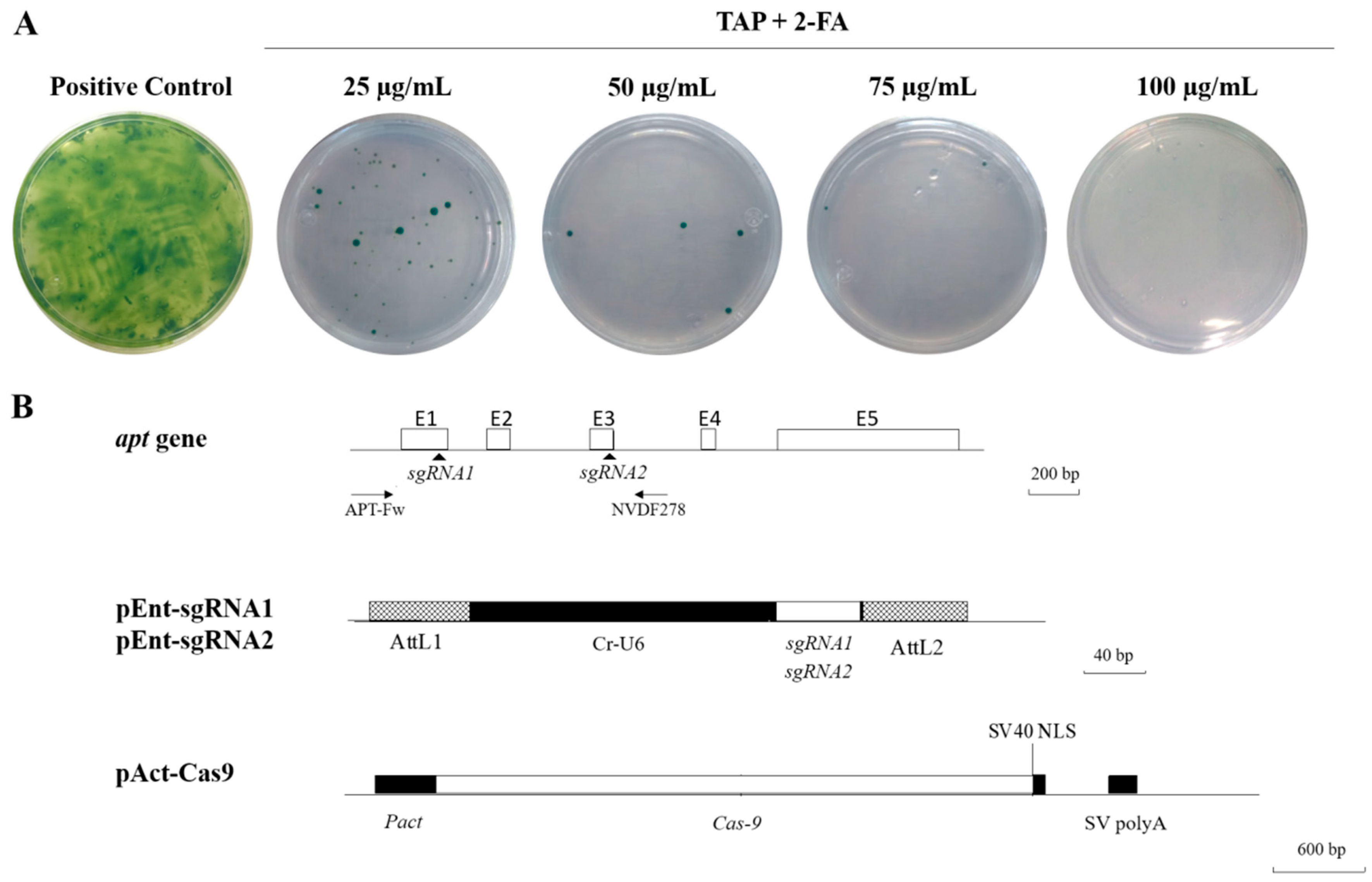{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. RNA Guides and Cas9 Plasmids
4.3. Chlamydomonas reinhardtii Nuclear Transformation with Glass Beads
4.4. Chlamydomonas reinhardtii Nuclear Transformation with Particle Bombardment
4.5. PCR Amplification of APT Gene
4.6. Sequence Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA—Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhou, H.; Bi, H.; Fromm, M.; Yang, B.; Weeks, D.P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Shin, S.E.; Lim, J.M.; Koh, H.G.; Kim, E.K.; Kang, N.K.; Jeon, S.; Kwon, S.; Shin, W.S.; Lee, B.; Hwangbo, K.; et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Greiner, A.; Kelterborn, S.; Evers, H.; Kreimer, G.; Sizova, I.; Hegemann, P. Targeting of Photoreceptor Genes in Chlamydomonas reinhardtii via Zinc-finger Nucleases and CRISPR/Cas9. Plant Cell 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Yu, J.; Jeong, J.; Sim, S.J.; Bae, S.; Jin, E.S. Photoautotrophic production of macular pigment in a Chlamydomonas reinhardtii strain generated by using DNA-free CRISPR-Cas9 RNP-mediated mutagenesis. Biotechnol. Bioeng. 2018, 115, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Kim, D.H.; Jeong, J.; Sim, S.J.; Melis, A.; Kim, J.-S.; Jin, E.; Bae, S. DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci. Rep. 2016, 6, 30620. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR—Cas9 Structures and Mechanisms. Annu. Rev. Biophys 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Sasso, S.; Stibor, H.; Mittag, M.; Grossman, A.R. From molecular manipulation of domesticated Chlamydomonas reinhardtii to survival in nature. Elife 2018, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Scranton, M.A.; Ostrand, J.T.; Fields, F.J.; Mayfield, S.P. Chlamydomonas as a model for biofuels and bio-products production. Plant J. 2015, 82, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozet, P.; Navarro, F.; Willmund, F.; Mehrshahi, P.; Bakowski, K.; Lauersen, K.; Pérez-Pérez, M.; Auroy, P.; Gorchs Rovira, A.; Sauret-Gueto, S.; et al. Birth of a photosynthetic chassis: A MoClo toolkit enabling synthetic biology in the microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 2018, 7, 2074–2086. [Google Scholar] [CrossRef]
- Li, X.; Zhang, R.; Patena, W.; Gang, S.S.; Blum, S.R.; Ivanova, N.; Yue, R.; Robertson, J.M.; Lefebvre, P.A.; Fitz-Gibbon, S.T.; et al. An Indexed, Mapped Mutant Library Enables Reverse Genetics Studies of Biological Processes in Chlamydomonas reinhardtii. Plant Cell 2016, 28, 367–387. [Google Scholar] [CrossRef]
- Jiang, W.; Brueggeman, A.J.; Horken, K.M.; Plucinak, T.M.; Weeks, D.P. Successful transient expression of Cas9 and single guide RNA genes in Chlamydomonas reinhardtii. Eukaryot. Cell 2014, 13, 1465–1469. [Google Scholar] [CrossRef]
- Jiang, W.Z.; Weeks, D.P. A gene-within-a-gene Cas9/sgRNA hybrid construct enables gene editing and gene replacement strategies in Chlamydomonas reinhardtii. Algal Res. 2017, 26, 474–480. [Google Scholar] [CrossRef]
- Ferenczi, A.; Pyott, D.E.; Xipnitou, A.; Molnar, A. Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Natl. Acad. Sci. USA 2017, 12, 201710597. [Google Scholar]
- Ashihara, H.; Stasolla, C.; Fujimura, T.; Crozier, A. Purine salvage in plants. Phytochemistry 2018, 147, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Schaff, D.A. The adenine phosphoribosyltransferase (APRT) selectable marker system. Plant Sci. 1994, 101, 3–9. [Google Scholar] [CrossRef]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ge, X.; Yang, F.; Zhang, L.; Zheng, J.; Tan, X.; Jin, Z.B.; Qu, J.; Gu, F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef]
- Shin, H.Y.; Wang, C.; Lee, H.K.; Yoo, K.H.; Zeng, X.; Kuhns, T.; Yang, C.M.; Mohr, T.; Liu, C.; Hennighausen, L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, 5–12. [Google Scholar] [CrossRef]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Thyme, S.B.; Akhmetova, L.; Montague, T.G.; Valen, E.; Schier, A.F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Kim, K.S.; Kustu, S.; Inwood, W. Natural history of transposition in the green alga Chlamydomonas reinhardtii: Use of the AMT4 locus as an experimental system. Genetics 2006, 173, 2005–2019. [Google Scholar] [CrossRef]
- Ono, R.; Ishii, M.; Fujihara, Y.; Kitazawa, M.; Usami, T.; Kaneko-Ishino, T.; Kanno, J.; Ikawa, M.; Ishino, F. Double strand break repair by capture of retrotransposon sequences and reverse-transcribed spliced mRNA sequences in mouse zygotes. Sci. Rep. 2015, 5, 12281. [Google Scholar] [CrossRef] [Green Version]
- Trouiller, B.; Charlot, F.; Choinard, S.; Schaefer, D.G.; Nogué, F. Comparison of gene targeting efficiencies in two mosses suggests that it is a conserved feature of Bryophyte transformation. Biotechnol. Lett. 2007, 29, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Collonnier, C.; Epert, A.; Mara, K.; Maclot, F.; Guyon-Debast, A.; Charlot, F.; White, C.; Schaefer, D.G.; Nogué, F. CRISPR-Cas9-mediated efficient directed mutagenesis and RAD51-dependent and RAD51-independent gene targeting in the moss Physcomitrella patens. Plant Biotechnol. J. 2017, 15, 122–131. [Google Scholar] [CrossRef]
- Miao, J.; Guo, D.; Zhang, J.; Huang, Q.; Qin, G.; Zhang, X.; Wan, J.; Gu, H.; Qu, L.-J. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 2013, 23, 1233. [Google Scholar] [CrossRef]
- Hamada, H.; Liu, Y.; Nagira, Y.; Miki, R.; Taoka, N.; Imai, R. Biolistic-delivery-based transient CRISPR/Cas9 expression enables in planta genome editing in wheat. Sci. Rep. 2018, 8, 14422. [Google Scholar] [CrossRef]
- Svitashev, S.; Young, J.K.; Schwartz, C.; Gao, H.; Falco, S.C.; Cigan, A.M. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiol. 2015, 169, 931–945. [Google Scholar] [CrossRef]
- Coll, J.M. Review. Methodologies for transferring DNA into eukaryotic microalgae. Spanish J. Agric. Res. 2006, 4, 316–330. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Li, T.; Zhang, Y.; Wang, Y.; Zhao, Q.; Liu, J.; Zhang, H.; Liu, C.; Ran, Y.; et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 2017, 8, 1–5. [Google Scholar] [CrossRef]
- Pawluk, A. CRISPR: No Sign of Slowing Down. Cell 2018, 174, 1039–1041. [Google Scholar] [CrossRef]
- Belshaw, N.; Grouneva, I.; Aram, L.; Gal, A.; Hopes, A.; Mock, T. Efficient CRISPR/Cas-mediated homologous recombination in the model diatom Thalassiosira pseudonana. bioRxiv 2017, 215582. [Google Scholar] [CrossRef]
- Nymark, M.; Sharma, A.K.; Sparstad, T.; Bones, A.M.; Winge, P. A CRISPR/Cas9 system adapted for gene editing in marine algae. Sci. Rep. 2016, 6, 6–11. [Google Scholar] [CrossRef]
- Poliner, E.; Takeuchi, T.; Du, Z.Y.; Benning, C.; Farré, E.M. Nontransgenic Marker-Free Gene Disruption by an Episomal CRISPR System in the Oleaginous Microalga, Nannochloropsis oceanica CCMP1779. ACS Synth. Biol. 2018, 7. [Google Scholar] [CrossRef]
- Gorman, D.S.; Levine, R.P. Cytochrome f and plastocyanin: Their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1965, 54, 1665–1669. [Google Scholar] [CrossRef]
- Jakab, G.; Mougin, A.; Kis, M.; Pollák, T.; Antal, M.; Branlant, C.; Solymosy, F. Chlamydomonas U2, U4 and U6 snRNAs. An evolutionary conserved putative third interaction between U4 and U6 snRNAs which has a counterpart in the U4atac-U6atac snRNA duplex. Biochimie 1997, 79, 387–395. [Google Scholar] [CrossRef]
- Guzmán-Zapata, D.; Macedo-Osorio, K.S.; Almaraz-Delgado, A.L.; Durán-Figueroa, N.; Badillo-Corona, J.A. Production of recombinant proteins in the chloroplast of the green alga Chlamydomonas reinhardtii. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; ISBN 9781627032384. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán-Zapata, D.; Sandoval-Vargas, J.M.; Macedo-Osorio, K.S.; Salgado-Manjarrez, E.; Castrejón-Flores, J.L.; Oliver-Salvador, M.d.C.; Durán-Figueroa, N.V.; Nogué, F.; Badillo-Corona, J.A. Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module. Int. J. Mol. Sci. 2019, 20, 1247. https://doi.org/10.3390/ijms20051247
Guzmán-Zapata D, Sandoval-Vargas JM, Macedo-Osorio KS, Salgado-Manjarrez E, Castrejón-Flores JL, Oliver-Salvador MdC, Durán-Figueroa NV, Nogué F, Badillo-Corona JA. Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module. International Journal of Molecular Sciences. 2019; 20(5):1247. https://doi.org/10.3390/ijms20051247
Chicago/Turabian StyleGuzmán-Zapata, Daniel, José M. Sandoval-Vargas, Karla S. Macedo-Osorio, Edgar Salgado-Manjarrez, José L. Castrejón-Flores, María del Carmen Oliver-Salvador, Noé V. Durán-Figueroa, Fabien Nogué, and Jesús A. Badillo-Corona. 2019. "Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module" International Journal of Molecular Sciences 20, no. 5: 1247. https://doi.org/10.3390/ijms20051247
APA StyleGuzmán-Zapata, D., Sandoval-Vargas, J. M., Macedo-Osorio, K. S., Salgado-Manjarrez, E., Castrejón-Flores, J. L., Oliver-Salvador, M. d. C., Durán-Figueroa, N. V., Nogué, F., & Badillo-Corona, J. A. (2019). Efficient Editing of the Nuclear APT Reporter Gene in Chlamydomonas reinhardtii via Expression of a CRISPR-Cas9 Module. International Journal of Molecular Sciences, 20(5), 1247. https://doi.org/10.3390/ijms20051247