IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential


Abstract
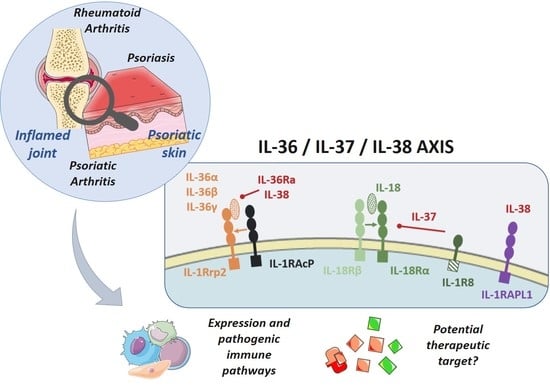
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. IL-36, IL-37, and IL-38: A Complex Group of Pro- and Anti-Inflammatory Cytokines
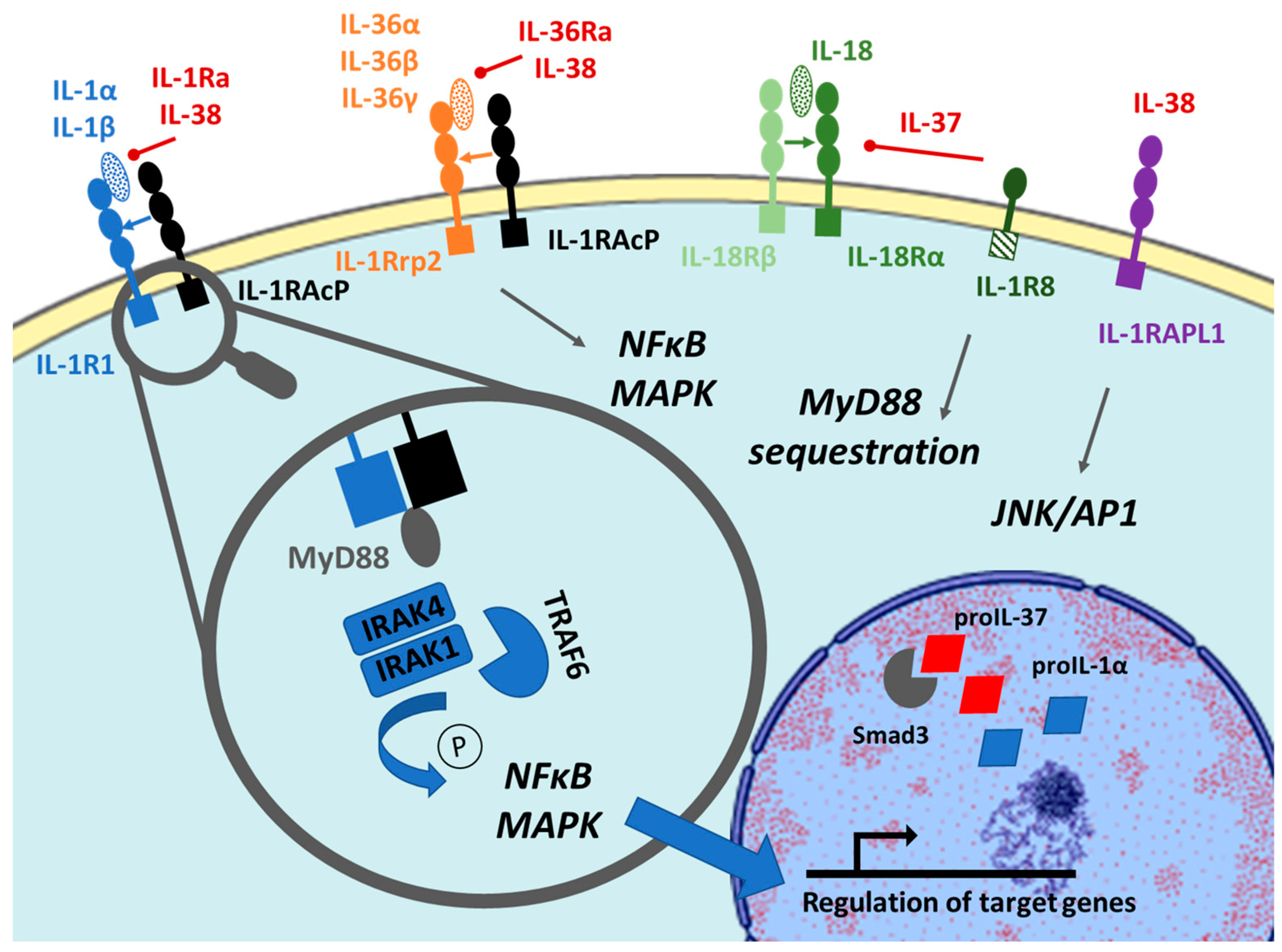
2.1. Maturation
2.1.1. IL-36 and IL-36Ra
2.1.2. IL-37
2.1.3. IL-38
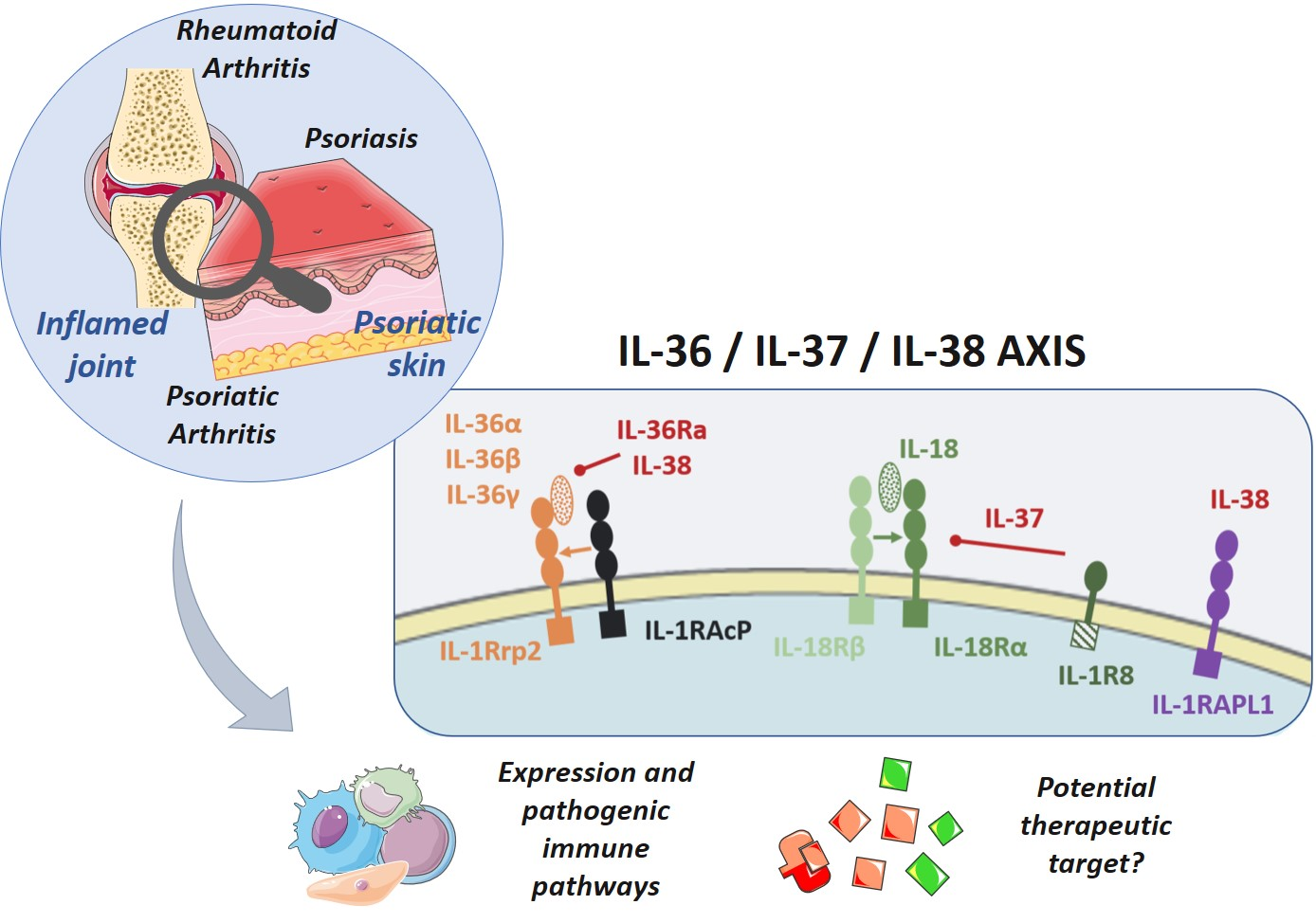
2.2. Receptors and Intracellular Signaling
2.2.1. IL-36 and IL-36Ra
2.2.2. IL-37
2.2.3. IL-38
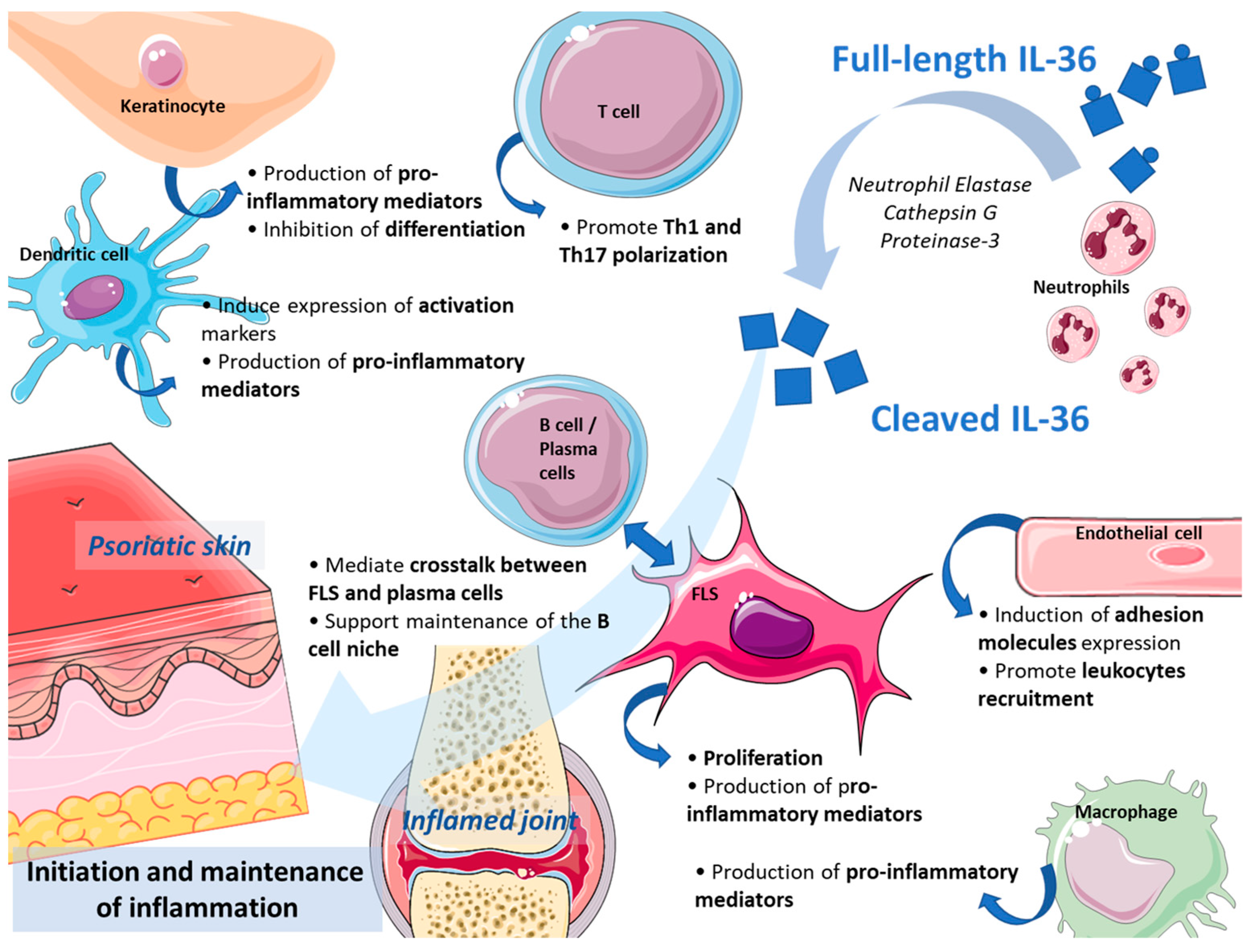
3. Expression and Role of IL-36 Cytokines in Inflamed Skin and Joints
3.1. Expression and Role in The inflamed Skin
3.2. Expression and Role in the Inflamed Joints
3.3. Are IL-36 Cytokines a Good Target in Skin- and Joint-Related Inflammation?
4. IL-37 and IL-38, Broad Inhibitors of Skin and Joint Inflammation
4.1. Anti-Inflammatory Role of IL-37 in Skin and Joints
4.2. Anti-Inflammatory Role of IL-38 in Skin and Joints
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| IL | Interleukin |
| IL-1Ra | IL-1 Receptor antagonist |
| PsA | Psoriatic Arthritis |
| RA | Rheumatoid Arthritis |
| CASPAR | ClASsification criteria for Psoriatic ARthritis |
| IL-36R | IL-36 Receptor |
| TLR | Toll-Like Receptor |
| PAMP | Pathogen-Associated Molecular Pattern |
| DAMP | Damage-Associated Molecular Pattern |
| NE | Neutrophil Elastase |
| NET | Neutrophil Extracellular Trap |
| MMP | Matrix Metalloproteinase |
| JNK | Jun N-terminal Kinase |
| IL-1RAPL1 | IL-1 Receptor Accessory Protein Like 1 |
| TIR | Toll/IL-1 Receptor |
| MyD88 | Myeloid Differentiation Primary Response Protein 88 |
| IL-1RAcP | IL-1 Receptor Accessory Protein |
| IL-1RL2 | IL-1 Receptor Like 2 |
| BP | Binding Protein |
| MAPK | Mitogen-Activated Protein Kinases |
| NFκB | Nuclear Factor-kappa B |
| IRAK | IL-1R-Associated Kinase |
| TRAF | TNF Receptor-associated Factor |
| PBMC | Peripheral Blood Mononuclear Cells |
| AP1 | Activator Protein 1 |
| LPS | Lipopolysaccharides |
| ds | Doubled-stranded |
| CD | Cluster of differentiation |
| MHC | Major Histocompatibility Complex |
| CCL1 | Chemokine (C-C motif) Ligand 1 |
| CXCL1 | Chemokine (C-X-C motif) Ligand 1 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| Th | T helper |
| VCAM | Vascular cell adhesion protein |
| ICAM | Intercellular Adhesion Molecule |
| DITRA | Deficiency of the Interleukin-36-Receptor Antagonist |
| CIA | Collagen-Induced Arthritis |
| FLS | Fibroblast-like Synoviocytes |
| ELS | Ectopic Lymphoid Structure |
| STAT | Signal Transducer and Activator of Transcription |
| GPP | Generalized Pustular Psoriasis |
| PPP | Palmoplantar pustulosis |
| S100A7 | S100 calcium-binding protein A7 |
| SCW | Streptococcal Cell Wall |
| STIA | Serum Transfer-Induced Arthritis |
| CRP | C-Reactive Protein |
| DMARD | Disease Modifying Anti-Rheumatic Drug |
References
- Kumar, S.; McDonnell, P.C.; Lehr, R.; Tierney, L.; Tzimas, M.N.; Griswold, D.E.; Capper, E.A.; Tal-Singer, R.; Wells, G.I.; Doyle, M.L.; et al. Identification and initial characterization of four novel members of the interleukin-1 family. J. Biol. Chem. 2000, 275, 10308–10314. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M. Global Epidemiology of Psoriasis: A Systematic Review of Incidence and Prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogdie, A.; Weiss, P. The Epidemiology of Psoriatic Arthritis. Rheum. Dis. Clin. N. Am. 2015, 41, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.-A.; Nerviani, A.; Gallo Afflitto, G.; Pitzalis, C. Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints. Int. J. Mol. Sci. 2018, 19, 530. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Carmona, L.; Wolfe, F.; Vos, T.; Williams, B.; Gabriel, S.; Lassere, M.; Johns, N.; et al. The global burden of rheumatoid arthritis: estimates from the Global Burden of Disease 2010 study. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef]
- Macleod, T.; Doble, R.; McGonagle, D.; Wasson, C.W.; Alase, A.; Stacey, M.; Wittmann, M. Neutrophil Elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 Receptor antagonist. Sci. Rep. 2016, 6, 24880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis. Drug Des. Devel. Ther. 2019, 13, 581–588. [Google Scholar] [CrossRef]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed]
- Ainscough, J.S.; Macleod, T.; McGonagle, D.; Brakefield, R.; Baron, J.M.; Alase, A.; Wittmann, M.; Stacey, M. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc. Natl. Acad. Sci. 2017, 114, E2748–E2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Frey, S.; Hueber, A.J. The novel interleukin-1 cytokine family members in inflammatory diseases. Curr. Opin. Rheumatol. 2017, 29, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Bulau, A.-M.; Nold, M.F.; Li, S.; Nold-Petry, C.A.; Fink, M.; Mansell, A.; Schwerd, T.; Hong, J.; Rubartelli, A.; Dinarello, C.A.; et al. Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 2650–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Hanning, C.R.; Brigham-Burke, M.R.; Rieman, D.J.; Lehr, R.; Khandekar, S.; Kirkpatrick, R.B.; Scott, G.F.; Lee, J.C.; Lynch, F.J.; et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 2002, 18, 61–71. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellisdon, A.M.; Nold-Petry, C.A.; D’Andrea, L.; Cho, S.X.; Lao, J.C.; Rudloff, I.; Ngo, D.; Lo, C.Y.; Soares da Costa, T.P.; Perugini, M.A.; et al. Homodimerization attenuates the anti-inflammatory activity of interleukin-37. Sci. Immunol. 2017, 2, eaaj1548. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.; Schlemmer, A.; Wittig, I.; Richter, F.; Putyrski, M.; Frank, A.-C.; Han, Y.; Jung, M.; Ernst, A.; Weigert, A.; et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell Biol. 2016. [Google Scholar] [CrossRef]
- Garraud, T.; Harel, M.; Boutet, M.-A.; Le Goff, B.; Blanchard, F. The enigmatic role of IL-38 in inflammatory diseases. Cytokine Growth Factor Rev. 2018, 39, 26–35. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Debets, R.; Timans, J.C.; Homey, B.; Zurawski, S.; Sana, T.R.; Lo, S.; Wagner, J.; Edwards, G.; Clifford, T.; Menon, S.; et al. Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J. Immunol. Baltim. Md 1950 2001, 167, 1440–1446. [Google Scholar]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef]
- Yi, G.; Ybe, J.A.; Saha, S.S.; Caviness, G.; Raymond, E.; Ganesan, R.; Mbow, M.L.; Kao, C.C. Structural and functional attributes of the Interleukin-36 receptor. J. Biol. Chem. 2016. [Google Scholar] [CrossRef]
- Zhou, L.; Todorovic, V.; Kakavas, S.; Sielaff, B.; Medina, L.; Wang, L.; Sadhukhan, R.; Stockmann, H.; Richardson, P.L.; DiGiammarino, E.; et al. Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation. J. Biol. Chem. 2018, 293, 403–411. [Google Scholar] [CrossRef]
- Bufler, P.; Azam, T.; Gamboni-Robertson, F.; Reznikov, L.L.; Kumar, S.; Dinarello, C.A.; Kim, S.-H. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 13723–13728. [Google Scholar] [CrossRef] [Green Version]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Riva, F.; Bonavita, E.; Barbati, E.; Muzio, M.; Mantovani, A.; Garlanda, C. TIR8/SIGIRR is an Interleukin-1 Receptor/Toll Like Receptor Family Member with Regulatory Functions in Inflammation and Immunity. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Dinarello, C.A. Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 2018, 281, 179–190. [Google Scholar] [CrossRef]
- Lin, H.; Ho, A.S.; Haley-Vicente, D.; Zhang, J.; Bernal-Fussell, J.; Pace, A.M.; Hansen, D.; Schweighofer, K.; Mize, N.K.; Ford, J.E. Cloning and characterization of IL-1HY2, a novel interleukin-1 family member. J. Biol. Chem. 2001, 276, 20597–20602. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Stoeckman, A.K.; Wu, G.; Boeckermann, A.N.; Azam, T.; Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M.; Hao, R.; Kalabokis, V.; et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. USA 2012, 109, 3001–3005. [Google Scholar] [CrossRef] [Green Version]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. Baltim. Md 1950 2011, 186, 2613–2622. [Google Scholar] [CrossRef]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schäfer, M.; Coyle, A.J.; Renauld, J.-C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Bachmann, M.; Scheiermann, P.; Härdle, L.; Pfeilschifter, J.; Mühl, H. IL-36γ/IL-1F9, an innate T-bet target in myeloid cells. J. Biol. Chem. 2012, 287, 41684–41696. [Google Scholar] [CrossRef]
- Carrier, Y.; Ma, H.-L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef]
- Lian, L.-H.; Milora, K.A.; Manupipatpong, K.K.; Jensen, L.E. The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of IL-36γ. J. Investig. Dermatol. 2012, 132, 1346–1353. [Google Scholar] [CrossRef]
- Gresnigt, M.S.; Rösler, B.; Jacobs, C.W.M.; Becker, K.L.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G.; Dinarello, C.A.; van de Veerdonk, F.L. The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur. J. Immunol. 2013, 43, 416–426. [Google Scholar] [CrossRef]
- Boutet, M.-A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.-C.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef]
- Friedrich, M.; Tillack, C.; Wollenberg, A.; Schauber, J.; Brand, S. IL-36γ sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Mattii, M.; Ayala, F.; Balato, N.; Filotico, R.; Lembo, S.; Schiattarella, M.; Patruno, C.; Marone, G.; Balato, A. The balance between pro- and anti-inflammatory cytokines is crucial in human allergic contact dermatitis pathogenesis: the role of IL-1 family members. Exp. Dermatol. 2013, 22, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; Cox, A.; McDonagh, A.J.G.; Nicklin, M.J.H.; di Giovine, F.S.; Timms, J.M.; Messenger, A.G.; Dimitropoulou, P.; Duff, G.W.; Cork, M.J. Genetic analysis of the interleukin-1 receptor antagonist and its homologue IL-1L1 in alopecia areata: strong severity association and possible gene interaction. Eur. J. Immunogenetics Off. J. Br. Soc. Histocompat. Immunogenetics 2002, 29, 25–30. [Google Scholar] [CrossRef]
- Heinemann, A.; He, Y.; Zimina, E.; Boerries, M.; Busch, H.; Chmel, N.; Kurz, T.; Bruckner-Tuderman, L.; Has, C. Induction of phenotype modifying cytokines by FERMT1 mutations. Hum. Mutat. 2011, 32, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Wang, W.; Yu, X.; Wu, C.; Jin, H. IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis. Int. J. Med. Sci. 2017, 14, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaff, C.M.; Marquardt, Y.; Fietkau, K.; Baron, J.M.; Lüscher, B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ohko, K.; Nakajima, K.; Kataoka, S.; Takaishi, M.; Sano, S. IL-36 signaling is essential for psoriatic inflammation through the augmentation of innate immune responses. J. Investig. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, D.; Martin, P.; Flacher, V.; Sun, Y.; Jarrossay, D.; Brembilla, N.; Mueller, C.; Arnett, H.A.; Palmer, G.; Towne, J.; et al. Interleukin-36 potently stimulates human M2 macrophages, Langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 2016, 84, 88–98. [Google Scholar] [CrossRef]
- Cai, Y.; Fleming, C.; Yan, J. New insights of T cells in the pathogenesis of psoriasis. Cell. Mol. Immunol. 2012, 9, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penha, R.; Higgins, J.; Mutamba, S.; Barrow, P.; Mahida, Y.; Foster, N. IL-36 receptor is expressed by human blood and intestinal T lymphocytes and is dose-dependently activated via IL-36β and induces CD4+ lymphocyte proliferation. Cytokine 2016, 85, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Martin, P.; Lamacchia, C.; Strebel, D.; Rodriguez, E.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ronchi, F.; et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 2012, 120, 3478–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgewood, C.; Fearnley, G.W.; Berekmeri, A.; Laws, P.; Macleod, T.; Ponnambalam, S.; Stacey, M.; Graham, A.; Wittmann, M. IL-36γ Is a Strong Inducer of IL-23 in Psoriatic Cells and Activates Angiogenesis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germán, B.; Wei, R.; Hener, P.; Martins, C.; Ye, T.; Gottwick, C.; Yang, J.; Seneschal, J.; Boniface, K.; Li, M. Disrupting the IL-36 and IL-23/IL-17 loop underlies the efficacy of calcipotriol and corticosteroid therapy for psoriasis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Rahman, P.; Sun, S.; Peddle, L.; Snelgrove, T.; Melay, W.; Greenwood, C.; Gladman, D. Association between the interleukin-1 family gene cluster and psoriatic arthritis. Arthritis Rheum. 2006, 54, 2321–2325. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef]
- Milora, K.A.; Fu, H.; Dubaz, O.; Jensen, L.E. Unprocessed Interleukin-36α Regulates Psoriasis-Like Skin Inflammation in Cooperation with Interleukin-1. J. Investig. Dermatol. 2015, 135, 2992–3000. [Google Scholar] [CrossRef]
- Alvarez, P.; Jensen, L.E. Imiquimod Treatment Causes Systemic Disease in Mice Resembling Generalized Pustular Psoriasis in an IL-1 and IL-36 Dependent Manner. Mediators Inflamm. 2016, 2016, 6756138. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef]
- Kivelevitch, D.; Frieder, J.; Watson, I.; Paek, S.Y.; Menter, M.A. Pharmacotherapeutic approaches for treating psoriasis in difficult-to-treat areas. Expert Opin. Pharmacother. 2018, 19, 561–575. [Google Scholar] [CrossRef]
- Mahil, S.K.; Catapano, M.; Di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med. 2017, 9, 2514. [Google Scholar] [CrossRef]
- Frey, S.; Derer, A.; Messbacher, M.-E.; Baeten, D.L.P.; Bugatti, S.; Montecucco, C.; Schett, G.; Hueber, A.J. The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis. 2013, 72, 1569–1574. [Google Scholar] [CrossRef]
- Magne, D.; Palmer, G.; Barton, J.L.; Mézin, F.; Talabot-Ayer, D.; Bas, S.; Duffy, T.; Noger, M.; Guerne, P.-A.; Nicklin, M.J.H.; et al. The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther. 2006, 8, R80. [Google Scholar] [CrossRef]
- Schmitt, V.; Hahn, M.; Kästele, V.; Wagner, O.; Wiendl, M.; Derer, A.; Taddeo, A.; Hahne, S.; Radbruch, A.; Jäck, H.-M.; et al. Interleukin-36 receptor mediates the crosstalk between plasma cells and synovial fibroblasts. Eur. J. Immunol. 2017, 47, 2101–2112. [Google Scholar] [CrossRef]
- Humby, F.; Bombardieri, M.; Manzo, A.; Kelly, S.; Blades, M.C.; Kirkham, B.; Spencer, J.; Pitzalis, C. Ectopic Lymphoid Structures Support Ongoing Production of Class-Switched Autoantibodies in Rheumatoid Synovium. PLoS Med. 2009, 6, e1. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, A.M.; Giraldo, N.A.; Petitprez, F.; Julie, C.; Lacroix, L.; Peschaud, F.; Emile, J.-F.; Marisa, L.; Fridman, W.H.; Storkus, W.J.; et al. Association of IL-36γ with tertiary lymphoid structures and inflammatory immune infiltrates in human colorectal cancer. Cancer Immunol. Immunother. 2018. [Google Scholar] [CrossRef]
- Harusato, A.; Abo, H.; Ngo, V.L.; Yi, S.W.; Mitsutake, K.; Osuka, S.; Kohlmeier, J.E.; Li, J.D.; Gewirtz, A.T.; Nusrat, A.; et al. IL-36γ signaling controls the induced regulatory T cell–Th9 cell balance via NFκB activation and STAT transcription factors. Mucosal Immunol. 2017, 10, 1455–1467. [Google Scholar] [CrossRef]
- Chowdhury, K.; Kumar, U.; Das, S.; Chaudhuri, J.; Kumar, P.; Kanjilal, M.; Ghosh, P.; Sircar, G.; Basyal, R.K.; Kanga, U.; et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Manzo, A.; Vitolo, B.; La Manna, M.P.; Giardina, G.; Sireci, G.; Dieli, F.; Montecucco, C.M.; et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatology 2015, 54, 2264–2272. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, F.; Guggino, G.; Ferrante, A.; Raimondo, S.; Bignone, R.; Rodolico, V.; Peralta, S.; Van Tok, M.; Cannizzaro, A.; Schinocca, C.; et al. Interleukin-9 Overexpression and Th9 Polarization Characterize the Inflamed Gut, the Synovial Tissue, and the Peripheral Blood of Patients With Psoriatic Arthritis: IL-9 IN PsA. Arthritis Rheumatol. 2016, 68, 1922–1931. [Google Scholar] [CrossRef]
- Derer, A.; Groetsch, B.; Harre, U.; Böhm, C.; Towne, J.; Schett, G.; Frey, S.; Hueber, A.J. Blockade of IL-36 Receptor Signaling Does Not Prevent from TNF-Induced Arthritis. PLoS ONE 2014, 9, e101954. [Google Scholar] [CrossRef] [PubMed]
- Lamacchia, C.; Palmer, G.; Rodriguez, E.; Martin, P.; Vigne, S.; Seemayer, C.A.; Talabot-Ayer, D.; Towne, J.E.; Gabay, C. The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis Res. Ther. 2013, 15, R38. [Google Scholar] [CrossRef]
- Dietrich, D.; Gabay, C. Inflammation: IL-36 has proinflammatory effects in skin but not in joints. Nat. Rev. Rheumatol. 2014, 10, 639–640. [Google Scholar] [CrossRef]
- Pitzalis, C.; Kelly, S.; Humby, F. New learnings on the pathophysiology of RA from synovial biopsies. Curr. Opin. Rheumatol. 2013, 25, 334–344. [Google Scholar] [CrossRef]
- Astorri, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Towards a stratified targeted approach with biologic treatments in rheumatoid arthritis: role of synovial pathobiology. Curr. Pharm. Des. 2015, 21, 2216–2224. [Google Scholar] [CrossRef]
- Khanskaya, I.; Pinkstaff, J.; Marino, M.H.; Savall, T.; Li, J. Marco Londei a Phase 1 Study of ANB019, an Anti-IL-36 Receptor Monoclonal Antibody, in Healthy Volunteers. Available online: https://www2.anaptysbio.com/wp-content/uploads/ANB019-Phase-1-Study-Poster-EAACI-2018.pdf (accessed on 29 May 2018).
- Tsai, Y.-C.; Tsai, T.-F. Anti-interleukin and interleukin therapies for psoriasis: current evidence and clinical usefulness. Ther. Adv. Musculoskelet. Dis. 2017, 9, 277–294. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, G.P.; Davidovich, P.B.; Sura-Trueba, S.; Belotcerkovskaya, E.; Henry, C.M.; Clancy, D.M.; Zinoveva, A.; Mametnabiev, T.; Garabadzhiu, A.V.; Martin, S.J. Identification of small-molecule elastase inhibitors as antagonists of IL-36 cytokine activation. FEBS Open Bio 2018, 8, 751–763. [Google Scholar] [CrossRef]
- Sullivan, G.P.; Henry, C.M.; Clancy, D.M.; Mametnabiev, T.; Belotcerkovskaya, E.; Davidovich, P.; Sura-Trueba, S.; Garabadzhiu, A.V.; Martin, S.J. Suppressing IL-36-driven inflammation using peptide pseudosubstrates for neutrophil proteases. Cell Death Dis. 2018, 9, 378. [Google Scholar] [CrossRef]
- Fitzgerald, O. Psoriatic arthritis synovial histopathology: commentary on the article by Kruithof and colleagues. Arthritis Res. Ther. 2005, 7, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Barajon, I.; Mantovani, A.; Garlanda, C. Regulatory Role of IL-1R8 in Immunity and Disease. Front. Immunol. 2016, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Keermann, M.; Kõks, S.; Reimann, E.; Abram, K.; Erm, T.; Silm, H.; Kingo, K. Expression of IL-36 family cytokines and IL-37 but not IL-38 is altered in psoriatic skin. J. Dermatol. Sci. 2015, 80, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Rouphael, N.; Duraisingham, S.; Romero-Steiner, S.; Presnell, S.; Davis, C.; Schmidt, D.S.; Johnson, S.E.; Milton, A.; Rajam, G.; et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014, 15, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Hu, Z.; Wei, X.; Wang, Z.; Guan, T.; Liu, N.; Liu, X.; Ye, N.; Deng, G.; Luo, C.; et al. IL-37 Ameliorates the Inflammatory Process in Psoriasis by Suppressing Proinflammatory Cytokine Production. J. Immunol. 2014, 192, 1815–1823. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Jiang, B.; Deng, J.; Du, J.; Xiong, W.; Guan, Y.; Wen, Z.; Huang, K.; Huang, Z. IL-37 Alleviates Rheumatoid Arthritis by Suppressing IL-17 and IL-17-Triggering Cytokine Production and Limiting Th17 Cell Proliferation. J. Immunol. Baltim. Md 1950 2015, 194, 5110–5119. [Google Scholar] [CrossRef]
- Cavalli, G.; Koenders, M.; Kalabokis, V.; Kim, J.; Tan, A.C.; Garlanda, C.; Mantovani, A.; Dagna, L.; Joosten, L.A.B.; Dinarello, C.A. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatol. Oxf. Engl. 2016. [Google Scholar] [CrossRef]
- Tang, R.; Yi, J.; Yang, J.; Chen, Y.; Luo, W.; Dong, S.; Fei, J. Interleukin-37 inhibits osteoclastogenesis and alleviates inflammatory bone destruction. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Goldring, S.R. Bone and joint destruction in rheumatoid arthritis: what is really happening? J. Rheumatol. Suppl. 2002, 65, 44–48. [Google Scholar]
- Ragab, D.; Mobasher, S.; Shabaan, E. Elevated levels of IL-37 correlate with T cell activation status in rheumatoid arthritis patients. Cytokine 2019, 113, 305–310. [Google Scholar] [CrossRef]
- Batliwalla, F.M.; Li, W.; Ritchlin, C.T.; Xiao, X.; Brenner, M.; Laragione, T.; Shao, T.; Durham, R.; Kemshetti, S.; Schwarz, E.; et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol. Med. Camb. Mass 2005, 11, 21–29. [Google Scholar]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Yuan, X.L.; Li, Y.; Pan, X.H.; Zhou, M.; Gao, Q.Y.; Li, M.C. Production of recombinant human interleukin-38 and its inhibitory effect on the expression of proinflammatory cytokines in THP-1 cells. Mol. Biol. 2016, 50, 466–473. [Google Scholar] [CrossRef]
- Boutet, M.-A.; Najm, A.; Bart, G.; Brion, R.; Touchais, S.; Trichet, V.; Layrolle, P.; Gabay, C.; Palmer, G.; Blanchard, F.; et al. IL-38 overexpression induces anti-inflammatory effects in mice arthritis models and in human macrophages in vitro. Ann. Rheum. Dis. 2017. [Google Scholar] [CrossRef]
- Shibata, A.; Sugiura, K.; Furuta, Y.; Mukumoto, Y.; Kaminuma, O.; Akiyama, M. Toll-like receptor 4 antagonist TAK-242 inhibits autoinflammatory symptoms in DITRA. J. Autoimmun. 2017. [Google Scholar] [CrossRef]
- Rudloff, I.; Godsell, J.; Nold-Petry, C.A.; Harris, J.; Hoi, A.; Morand, E.F.; Nold, M.F. Brief Report: Interleukin-38 Exerts Antiinflammatory Functions and Is Associated with Disease Activity in Systemic Lupus Erythematosus. Arthritis Rheumatol. Hoboken NJ 2015, 67, 3219–3225. [Google Scholar] [CrossRef]
- Palomo, J.; Troccaz, S.; Talabot-Ayer, D.; Rodriguez, E.; Palmer, G. The severity of imiquimod-induced mouse skin inflammation is independent of endogenous IL-38 expression. PLOS ONE 2018, 13, e0194667. [Google Scholar] [CrossRef]
- Chu, M.; Tam, L.S.; Zhu, J.; Jiao, D.; Liu, D.H.; Cai, Z.; Dong, J.; Kai Lam, C.W.; Wong, C.K. In vivo anti-inflammatory activities of novel cytokine IL-38 in Murphy Roths Large (MRL)/lpr mice. Immunobiology 2016. [Google Scholar] [CrossRef]
- Takenaka, S.; Kaieda, S.; Kawayama, T.; Matsuoka, M.; Kaku, Y.; Kinoshita, T.; Sakazaki, Y.; Okamoto, M.; Tominaga, M.; Kanesaki, K.; et al. IL-38: A new factor in rheumatoid arthritis. Biochem. Biophys. Rep. 2015, 4, 386–391. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-D.; Su, L.-C.; He, C.-S.; Huang, A.-F. Plasma interleukin-38 in patients with rheumatoid arthritis. Int. Immunopharmacol. 2018, 65, 1–7. [Google Scholar] [CrossRef]
- Chou, C.-T.; Timms, A.E.; Wei, J.C.C.; Tsai, W.C.; Wordsworth, B.P.; Brown, M.A. Replication of association of IL1 gene complex members with ankylosing spondylitis in Taiwanese Chinese. Ann. Rheum. Dis. 2006, 65, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Li, C.; Lin, Z.M.; Huang, J.X.; Wei, Q.J.; Wang, X.W.; Xie, Y.Y.; Liao, Z.T.; Chao, S.Y.; Gu, J.R. Association of IL-1 gene complex members with ankylosing spondylitis in Chinese Han population. Int. J. Immunogenet. 2010, 37, 33–37. [Google Scholar] [CrossRef]
- Dehghan, A.; Dupuis, J.; Barbalic, M.; Bis, J.C.; Eiriksdottir, G.; Lu, C.; Pellikka, N.; Wallaschofski, H.; Kettunen, J.; Henneman, P.; et al. Meta-analysis of genome-wide association studies in >80,000 subjects identifies multiple loci for C-reactive protein levels. Circulation 2011, 123, 731–738. [Google Scholar] [CrossRef]
- Jung, M.Y.; Kang, S.W.; Kim, S.K.; Kim, H.-J.; Yun, D.H.; Yim, S.-V.; Hong, S.J.; Chung, J.-H. The interleukin-1 family gene polymorphisms in Korean patients with rheumatoid arthritis. Scand. J. Rheumatol. 2010, 39, 190–196. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutet, M.-A.; Nerviani, A.; Pitzalis, C. IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 1257. https://doi.org/10.3390/ijms20061257
Boutet M-A, Nerviani A, Pitzalis C. IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential. International Journal of Molecular Sciences. 2019; 20(6):1257. https://doi.org/10.3390/ijms20061257
Chicago/Turabian StyleBoutet, Marie-Astrid, Alessandra Nerviani, and Costantino Pitzalis. 2019. "IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential" International Journal of Molecular Sciences 20, no. 6: 1257. https://doi.org/10.3390/ijms20061257
APA StyleBoutet, M. -A., Nerviani, A., & Pitzalis, C. (2019). IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential. International Journal of Molecular Sciences, 20(6), 1257. https://doi.org/10.3390/ijms20061257