Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models

 , , ,
, , ,
Abstract
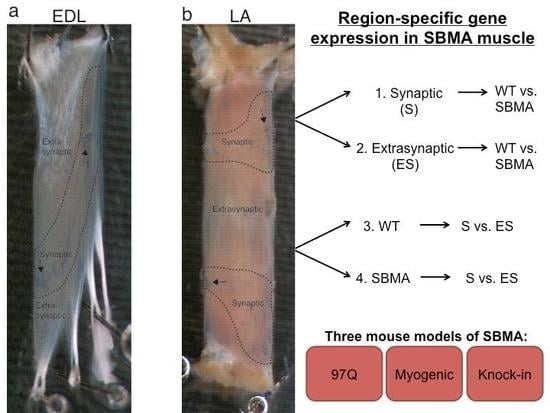
:
1. Introduction
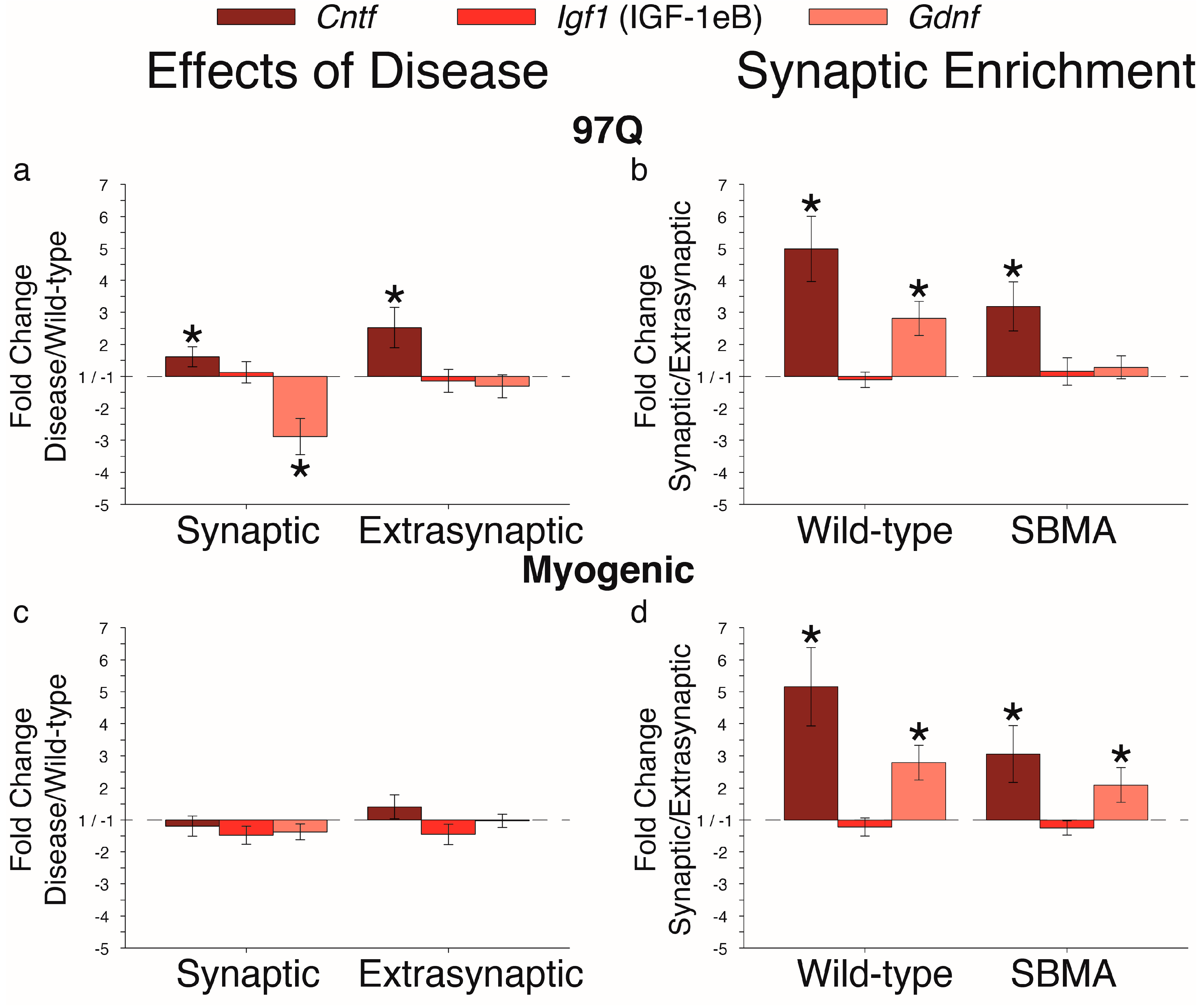
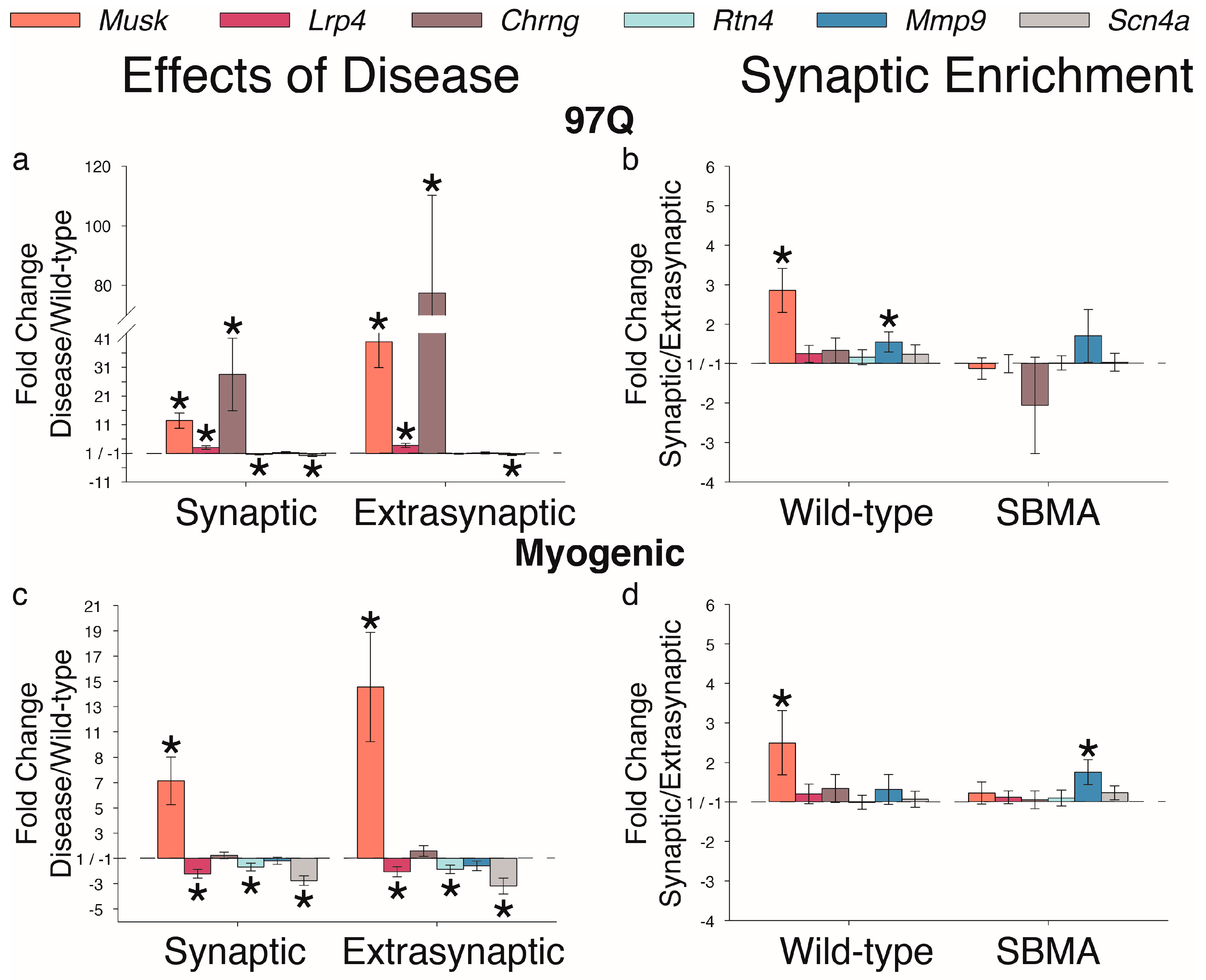
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. AR97Q Model
4.3. Myogenic Model
4.4. AR113Q Knock-In (KI) Model
4.5. Muscle Dissection
4.6. Quantitative Reverse-Transcription PCR
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katsuno, M.; Tanaka, F.; Adachi, H.; Banno, H.; Suzuki, K.; Watanabe, H.; Sobue, G. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog. Neurobiol. 2012, 99, 246–256. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Adachi, H.; Kume, A.; Li, M.; Nakagomi, Y.; Niwa, H.; Sang, C.; Kobayashi, Y.; Doyu, M.; Sobue, G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002, 35, 843–854. [Google Scholar] [CrossRef]
- Takeyama, K.; Ito, S.; Yamamoto, A.; Tanimoto, H.; Furutani, T.; Kanuka, H.; Miura, M.; Tabata, T.; Kato, S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 2002, 35, 855–864. [Google Scholar] [CrossRef]
- Monks, D.A.; Johansen, J.A.; Mo, K.; Rao, P.; Eagleson, B.; Yu, Z.; Lieberman, A.P.; Breedlove, S.M.; Jordan, C.L. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18259–18264. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Dadgar, N.; Albertelli, M.; Gruis, K.; Jordan, C.; Robins, D.M.; Lieberman, A.P. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Investig. 2006, 116, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- Chevalier-Larsen, E.S.; O’Brien, C.J.; Wang, H.; Jenkins, S.C.; Holder, L.; Lieberman, A.P.; Merry, D.E. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J. Neurosci. 2004, 24, 4778–4786. [Google Scholar] [CrossRef]
- Johansen, J.A.; Yu, Z.; Mo, K.; Monks, D.A.; Lieberman, A.P.; Breedlove, S.M.; Jordan, C.L. Recovery of function in a myogenic mouse model of spinal bulbar muscular atrophy. Neurobiol. Dis. 2009, 34, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; Ling, S.C.; Guo, L.T.; Hung, G.; Tsunemi, T.; Ly, L.; Tokunaga, S.; Lopez, E.; Sopher, B.L.; Bennett, C.F.; et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014, 82, 295–307. [Google Scholar] [CrossRef]
- Oki, K.; Halievski, K.; Vicente, L.; Xu, Y.; Zeolla, D.; Poort, J.; Katsuno, M.; Adachi, H.; Sobue, G.; Wiseman, R.W.; et al. Contractile dysfunction in muscle may underlie androgen-dependent motor dysfunction in spinal bulbar muscular atrophy. J. Appl. Physiol. (1985) 2015, 118, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Oki, K.; Wiseman, R.W.; Breedlove, S.M.; Jordan, C.L. Androgen receptors in muscle fibers induce rapid loss of force but not mass: Implications for spinal bulbar muscular atrophy. Muscle Nerve 2013, 47, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.Q.; Poort, J.L.; Baqri, R.M.; Lieberman, A.P.; Breedlove, S.M.; Miller, K.E.; Jordan, C.L. Impaired motoneuronal retrograde transport in two models of SBMA implicates two sites of androgen action. Hum. Mol. Genet. 2011, 20, 4475–4490. [Google Scholar] [CrossRef] [Green Version]
- Halievski, K.; Kemp, M.Q.; Breedlove, S.M.; Miller, K.E.; Jordan, C.L. Non-Cell-Autonomous Regulation of Retrograde Motoneuronal Axonal Transport in an SBMA Mouse Model. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Halievski, K.; Henley, C.; Atchison, W.D.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Defects in Neuromuscular Transmission May Underlie Motor Dysfunction in Spinal and Bulbar Muscular Atrophy. J. Neurosci. 2016, 36, 5094–5106. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Halievski, K.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Pre-clinical symptoms of SBMA may not be androgen-dependent: Implications from two SBMA mouse models. Hum. Mol. Genet. 2018, 27, 2425–2442. [Google Scholar] [CrossRef]
- Halievski, K.; Mo, K.; Westwood, J.T.; Monks, D.A. Transcriptional profile of muscle following acute induction of symptoms in a mouse model of Kennedy’s disease/spinobulbar muscular atrophy. PLoS ONE 2015, 10, e0118120. [Google Scholar] [CrossRef]
- Mo, K.; Razak, Z.; Rao, P.; Yu, Z.; Adachi, H.; Katsuno, M.; Sobue, G.; Lieberman, A.P.; Westwood, J.T.; Monks, D.A. Microarray analysis of gene expression by skeletal muscle of three mouse models of Kennedy disease/spinal bulbar muscular atrophy. PLoS ONE 2010, 5, e12922. [Google Scholar] [CrossRef]
- Giorgetti, E.; Yu, Z.; Chua, J.P.; Shimamura, R.; Zhao, L.; Zhu, F.; Venneti, S.; Pennuto, M.; Guan, Y.; Hung, G.; et al. Rescue of Metabolic Alterations in AR113Q Skeletal Muscle by Peripheral Androgen Receptor Gene Silencing. Cell Rep. 2016, 17, 125–136. [Google Scholar] [CrossRef]
- Sanes, J.R.; Johnson, Y.R.; Kotzbauer, P.T.; Mudd, J.; Hanley, T.; Martinou, J.C.; Merlie, J.P. Selective expression of an acetylcholine receptor-lacZ transgene in synaptic nuclei of adult muscle fibers. Development 1991, 113, 1181–1191. [Google Scholar]
- Monks, D.A.; O’Bryant, E.L.; Jordan, C.L. Androgen receptor immunoreactivity in skeletal muscle: Enrichment at the neuromuscular junction. J. Comp. Neurol. 2004, 473, 59–72. [Google Scholar] [CrossRef]
- Johansen, J.A.; Breedlove, S.M.; Jordan, C.L. Androgen receptor expression in the levator ani muscle of male mice. J. Neuroendocrinol. 2007, 19, 823–826. [Google Scholar] [CrossRef]
- Halievski, K.; Henley, C.L.; Domino, L.; Poort, J.E.; Fu, M.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Androgen-dependent loss of muscle BDNF mRNA in two mouse models of SBMA. Exp. Neurol. 2015, 269, 224–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Mitsuma, N.; Inukai, A.; Ito, Y.; Li, M.; Mitsuma, T.; Sobue, G. Expression of GDNF and GDNFR-alpha mRNAs in muscles of patients with motor neuron diseases. Neurochem. Res. 1999, 24, 785–790. [Google Scholar] [CrossRef]
- Palazzolo, I.; Stack, C.; Kong, L.; Musaro, A.; Adachi, H.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Sumner, C.J.; Fischbeck, K.H.; et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 2009, 63, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Parry, D.J.; Jasmin, B.J. BDNF rescues myosin heavy chain IIB muscle fibers after neonatal nerve injury. Am. J. Physiol. Cell Physiol. 2004, 287, C22–C29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, K.; Jasmin, B.J. BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 2006, 26, 5739–5749. [Google Scholar] [CrossRef]
- Kulakowski, S.A.; Parker, S.D.; Personius, K.E. Reduced TrkB expression results in precocious age-like changes in neuromuscular structure, neurotransmission, and muscle function. J. Appl. Physiol. (1985) 2011, 111, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Mantilla, C.B.; Zhan, W.Z.; Sieck, G.C. Neurotrophins improve neuromuscular transmission in the adult rat diaphragm. Muscle Nerve 2004, 29, 381–386. [Google Scholar] [CrossRef]
- Poort, J.E.; Rheuben, M.B.; Breedlove, S.M.; Jordan, C.L. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Giess, R.; Holtmann, B.; Braga, M.; Grimm, T.; Muller-Myhsok, B.; Toyka, K.V.; Sendtner, M. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: Potential impact of CNTF as a candidate modifier gene. Am. J. Hum. Genet. 2002, 70, 1277–1286. [Google Scholar] [CrossRef]
- Xu, J.; Gingras, K.M.; Bengston, L.; Di Marco, A.; Forger, N.G. Blockade of endogenous neurotrophic factors prevents the androgenic rescue of rat spinal motoneurons. J. Neurosci. 2001, 21, 4366–4372. [Google Scholar] [CrossRef]
- English, A.W. Cytokines, growth factors and sprouting at the neuromuscular junction. J. Neurocytol. 2003, 32, 943–960. [Google Scholar] [CrossRef]
- Chevrel, G.; Hohlfeld, R.; Sendtner, M. The role of neurotrophins in muscle under physiological and pathological conditions. Muscle Nerve 2006, 33, 462–476. [Google Scholar] [CrossRef]
- Pitts, E.V.; Potluri, S.; Hess, D.M.; Balice-Gordon, R.J. Neurotrophin and Trk-mediated signaling in the neuromuscular system. Int. Anesthesiol. Clin. 2006, 44, 21–76. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Je, H.S. Neurotrophic regulation of the development and function of the neuromuscular synapses. J. Neurocytol. 2003, 32, 931–941. [Google Scholar] [CrossRef]
- Valenzuela, D.M.; Stitt, T.N.; DiStefano, P.S.; Rojas, E.; Mattsson, K.; Compton, D.L.; Nunez, L.; Park, J.S.; Stark, J.L.; Gies, D.R.; et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: Expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 1995, 15, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.M.; Scott, M.O.; Potluri, S.; Pitts, E.V.; Cisterni, C.; Balice-Gordon, R.J. Localization of TrkC to Schwann cells and effects of neurotrophin-3 signaling at neuromuscular synapses. J. Comp. Neurol. 2007, 501, 465–482. [Google Scholar] [CrossRef]
- Brenner, H.R.; Witzemann, V.; Sakmann, B. Imprinting of acetylcholine receptor messenger RNA accumulation in mammalian neuromuscular synapses. Nature 1990, 344, 544–547. [Google Scholar] [CrossRef]
- Lohof, A.M.; Ip, N.Y.; Poo, M.M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 1993, 363, 350–353. [Google Scholar] [CrossRef]
- Sakuma, K.; Watanabe, K.; Sano, M.; Uramoto, I.; Nakano, H.; Li, Y.J.; Kaneda, S.; Sorimachi, Y.; Yoshimoto, K.; Yasuhara, M.; et al. A possible role for BDNF, NT-4 and TrkB in the spinal cord and muscle of rat subjected to mechanical overload, bupivacaine injection and axotomy. Brain Res. 2001, 907, 1–19. [Google Scholar] [CrossRef]
- Fernyhough, P.; Maeda, K.; Tomlinson, D.R. Brain-derived neurotrophic factor mRNA levels are up-regulated in hindlimb skeletal muscle of diabetic rats: Effect of denervation. Exp. Neurol. 1996, 141, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Veltri, K.; Li, S.; Bain, J.R.; Fahnestock, M. NGF, BDNF, NT-3, and GDNF mRNA expression in rat skeletal muscle following denervation and sensory protection. J. Neurotrauma 2004, 21, 1468–1478. [Google Scholar] [CrossRef]
- Funakoshi, H.; Frisen, J.; Barbany, G.; Timmusk, T.; Zachrisson, O.; Verge, V.M.; Persson, H. Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J. Cell Biol. 1993, 123, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N.; et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Physiol. Cell Physiol. 2012, 302, C141–C153. [Google Scholar] [CrossRef]
- Gonzalez, M.; Ruggiero, F.P.; Chang, Q.; Shi, Y.J.; Rich, M.M.; Kraner, S.; Balice-Gordon, R.J. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron 1999, 24, 567–583. [Google Scholar] [CrossRef]
- Rocchi, A.; Milioto, C.; Parodi, S.; Armirotti, A.; Borgia, D.; Pellegrini, M.; Urciuolo, A.; Molon, S.; Morbidoni, V.; Marabita, M.; et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016, 132, 127–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef]
- Herbst, R.; Avetisova, E.; Burden, S.J. Restoration of synapse formation in Musk mutant mice expressing a Musk/Trk chimeric receptor. Development 2002, 129, 5449–5460. [Google Scholar] [CrossRef] [Green Version]
- Wells, D.G.; McKechnie, B.A.; Kelkar, S.; Fallon, J.R. Neurotrophins regulate agrin-induced postsynaptic differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 1112–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Hayashi, K.; Ohi, T.; Imura, H. Level of nerve growth factor-like immunoreactivity in the lower limb muscles of muscular dystrophic mice. Biochem. Biophys. Res. Commun. 1979, 90, 130–134. [Google Scholar] [CrossRef]
- Capsoni, S.; Ruberti, F.; Di Daniel, E.; Cattaneo, A. Muscular dystrophy in adult and aged anti-NGF transgenic mice resembles an inclusion body myopathy. J. Neurosci. Res. 2000, 59, 553–560. [Google Scholar] [CrossRef]
- Lavasani, M.; Lu, A.; Peng, H.; Cummins, J.; Huard, J. Nerve growth factor improves the muscle regeneration capacity of muscle stem cells in dystrophic muscle. Hum. Gene Ther. 2006, 17, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Yamakuni, T.; Okabe, N.; Sakimura, K.; Takahashi, Y. Production of nerve growth factor in rat skeletal muscle. Neurosci. Lett. 1991, 132, 5–7. [Google Scholar] [CrossRef]
- Ko, C.P.; Robitaille, R. Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a020503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, D.; Vande Velde, C.; Robitaille, R. New perspectives on amyotrophic lateral sclerosis: The role of glial cells at the neuromuscular junction. J. Physiol. 2017, 595, 647–661. [Google Scholar] [CrossRef]
- Weydt, P.; Sagnelli, A.; Rosenbohm, A.; Fratta, P.; Pradat, P.F.; Ludolph, A.C.; Pareyson, D. Clinical Trials in Spinal and Bulbar Muscular Atrophy-Past, Present, and Future. J. Mol. Neurosci. 2016, 58, 379–387. [Google Scholar] [CrossRef]
- Johansen, J.A.; Troxell-Smith, S.M.; Yu, Z.; Mo, K.; Monks, D.A.; Lieberman, A.P.; Breedlove, S.M.; Jordan, C.L. Prenatal flutamide enhances survival in a myogenic mouse model of spinal bulbar muscular atrophy. Neuro-Degener. Dis. 2011, 8, 25–34. [Google Scholar] [CrossRef]
- Close, R. Properties of motor units in fast and slow skeletal muscles of the rat. J. Physiol. 1967, 193, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Edgerton, V.R.; Simpson, D.R. The intermediate muscle fiber of rats and guinea pigs. J. Histochem. Cytochem. 1969, 17, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, V.R.; Simpson, D.R. Dynamic and metabolic relationships in the rat extensor digitorum longus muscle. Exp. Neurol. 1971, 30, 374–376. [Google Scholar] [CrossRef]
- Hanzlikova, V.; Schiaffino, S.; Settembrini, P. Histochemical fiber types characteristics in the normal and the persistent levator ani muscle of the rat. Histochemie 1970, 22, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, V.; Gutmann, E. Effect of Foreign Innervation on the Androgen-Sensitive Levator Ani Muscle of the Rat. Z. Zellforsch 1972, 135, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
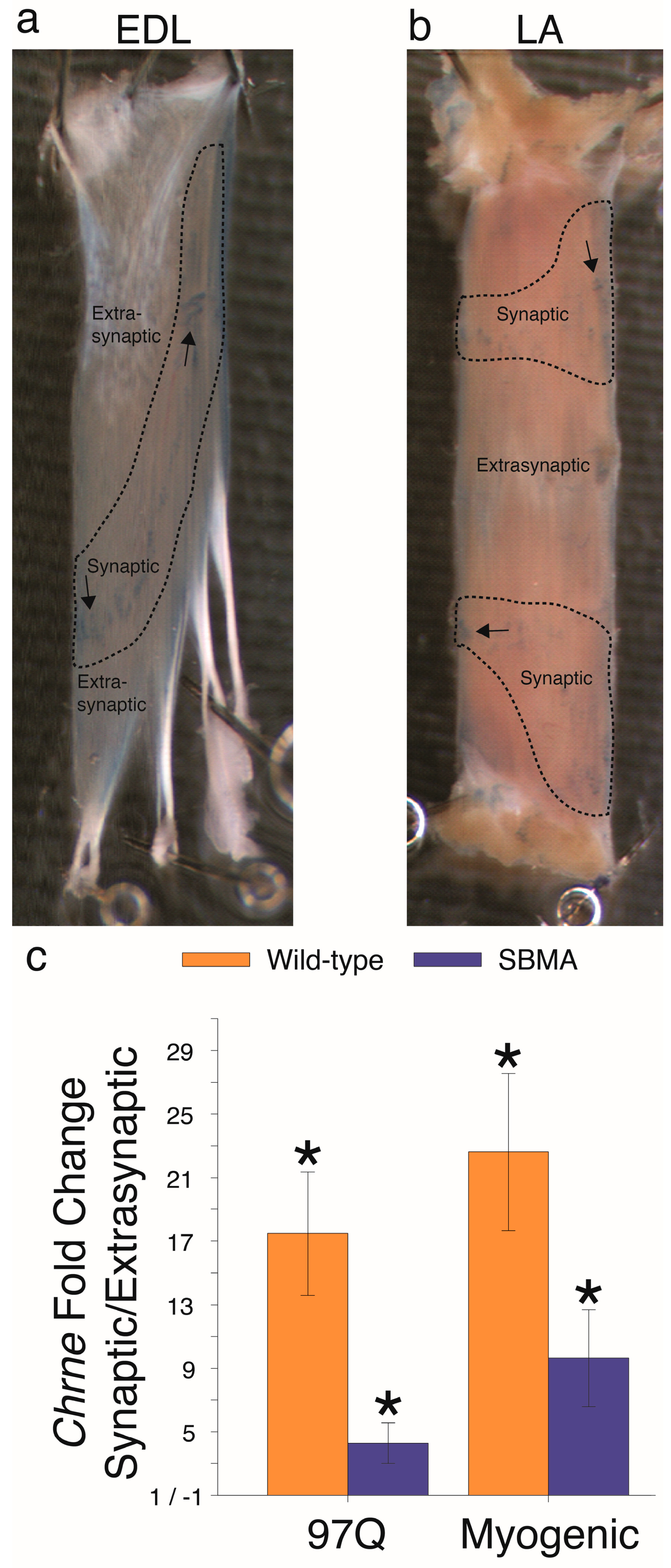{kind=link}
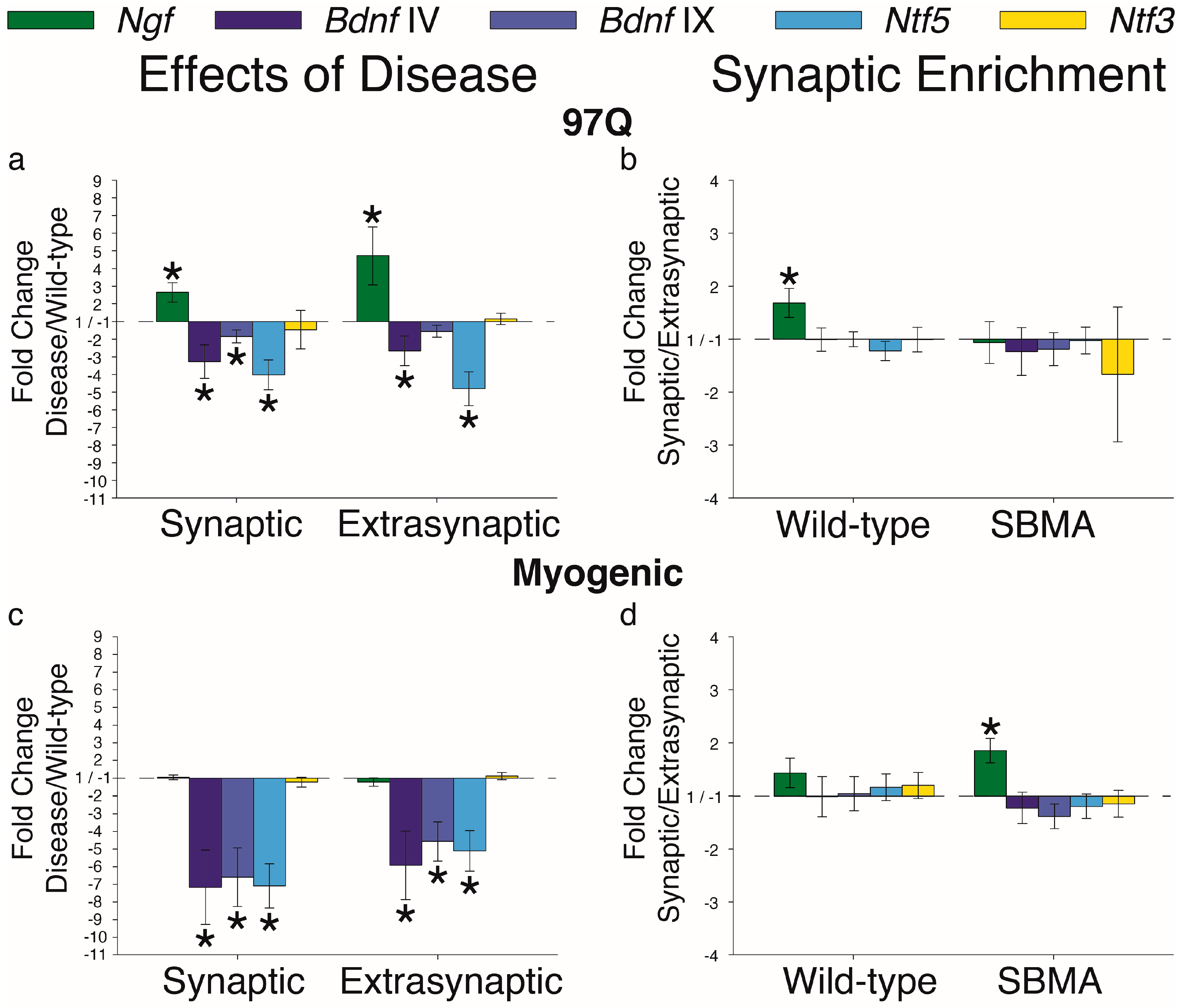{kind=link}
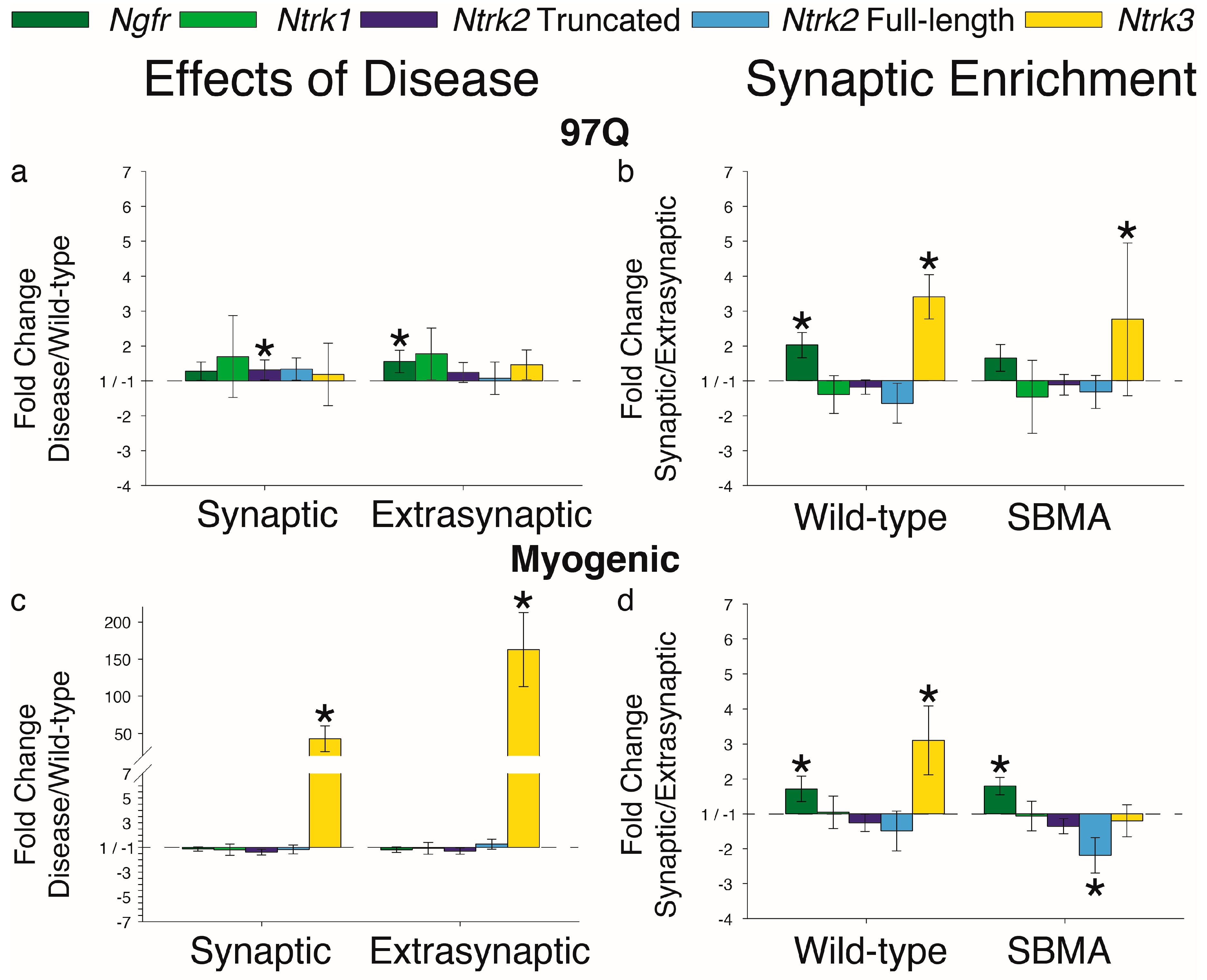{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chrne | Ngf | Bdnf (IV) | Bdnf (IX) | Ntf5 | Ntf3 | Ngfr | Ntrk1 | Ntrk2 (Truncated) | Ntrk2 (Full) | Ntrk3 | Cntf | Igf1 | Gdnf | Musk | Lrp4 | Chrng | Rtna | Mmp9 | Scn4a | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 97Q | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ * | ✔ | ✔ | ✔ * | ✔ | ✔ | ✔ | ✔ | ✔ | ||||||
| Myogenic | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ # | ✔ | ✔ | |||||||||||
| KI | ✔ | ✔ # | ✔ # | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Wild-Type | SBMA | |
|---|---|---|
| 97Q | ||
| Synaptic | 5 | 6 (5 for Gdnf; 4 for Rtn4 and Ntrk1) |
| Extrasynaptic | 5 | 6 (4 for Gdnf, Rtn4, and Ntrk1) |
| Myogenic | ||
| Synaptic | 6 | 7 |
| Extrasynaptic | 7 | 8 |
| Knock-in | ||
| Synaptic | 7 | 6 |
| Extrasynaptic | 7 | 6 (5 for Ntrk1) |
| Gene | Forward | Reverse | Calculated Efficiency | Concentration (nM) |
|---|---|---|---|---|
| Rn18s (18S) | GGACCAGAGCGAAAGCATTTG | GCCAGTCGGCATCGTTTATG | 1.90 | 100 |
| Chrne (AChR epsilon) | CTCTGCCAGAACCTGGGTG | TGTGCTCTCAGCCACAAAGT | 2.15 | 200 |
| Ngf | AGCTTTCTATACTGGCCGCA | TACGCCGATCAAAAACGCAG | 1.92 | 600 |
| Bdnf (exon IV) | CTCCGCCATGCAATTTCCAC | CGAGTCTTTGGTGGCCGATA | 1.74 | 200 |
| Bdnf (exon IX) | ACCATCCTTTTCCTTACTATGGTT | ATTCACGCTCTCCAGAGTCC | 1.98 | 200 |
| Ntf5 (NT-4) | TGAGCTGGCAGTATGCGAC | CAGCGCGTCTCGAAGAAGT | 2.03 | 600 |
| Ntf3 (NT-3) | TGGAGCCCCCTCCCTTATAC | AATGGCTGAGGACTTGTCGG | 2.23 | 100 |
| Ngfr (p75) | CGTGACCATCTCAGGCCTTT | GGTGCCCCTGTTACCTTCTC | 2.01 | 200 |
| Ntrk1 (TrkA) | ATATCTAGCCAGCCTGCACTTTGT | TGCTCATGCCAAAGTCTCCA | 2.15 | 600 |
| Ntrk2 (TrkB truncated) | CCATTGCCCTCTGCTAACCA | GAGATCTGAGGTGCTCTCGC | 2.08 | 600 |
| Ntrk2 (TrkB full length) | GGCAACTTCGGGAAAGGAGA | GTAAACCCCTCACCGCCTAC | 2.25 | 400 |
| Ntrk3 (TrkC) | ATGGAGCTCTACACGGGACT | GGTGAGCCGGTTACTTGACA | 2.40 | 600 |
| Cntf | TTTCACCCCGACTGAAGGTG | TTCTGTTCCAGAAGCGCCAT | 2.10 | 200 |
| Igf1 (IGF-1eb) | CCCGTCCCTATCGACAAACAA | TGGGAGGCTCCTCCTACATT | 2.00 | 100 |
| Gdnf | GCCACCATTAAAAGACTGAAAAGG | GCCTGCCGATTCCTCTCTCT | 1.91 | 600 |
| Musk | GCTGTTTGACACCCGCTACA | CTCCCACTCCATTGTTGGCTA | 1.97 | 400 |
| Lrp4 | GCATTGGTACTGCGATGGTG | CATAGGCGCACTGGAACTCT | 1.94 | 100 |
| Chrng (AChR gamma) | GGTTGGTGATGGGTATGGTCA | TGACATCAGGAAAGGCAGAGC | 2.06 | 200 |
| Rtn4 (Nogo-A) | ACTTACGTTGGTGCCTTGTTC | TGATCTATCTGCGCCTGATGC | 1.67 | 200 |
| Mmp9 | GCCGACTTTTGTGGTCTTCC | CTTCTCTCCCATCATCTGGGC | 2.01 | 200 |
| Scn4a (NaV1.4) | TGGGGGTCAACTTGTTTGCT | TCGAATCTCTCGGAGGTGGT | 2.09 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halievski, K.; Nath, S.R.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Lieberman, A.P.; Jordan, C.L. Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models. Int. J. Mol. Sci. 2019, 20, 1314. https://doi.org/10.3390/ijms20061314
Halievski K, Nath SR, Katsuno M, Adachi H, Sobue G, Breedlove SM, Lieberman AP, Jordan CL. Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models. International Journal of Molecular Sciences. 2019; 20(6):1314. https://doi.org/10.3390/ijms20061314
Chicago/Turabian StyleHalievski, Katherine, Samir R. Nath, Masahisa Katsuno, Hiroaki Adachi, Gen Sobue, S. Marc Breedlove, Andrew P. Lieberman, and Cynthia L. Jordan. 2019. "Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models" International Journal of Molecular Sciences 20, no. 6: 1314. https://doi.org/10.3390/ijms20061314
APA StyleHalievski, K., Nath, S. R., Katsuno, M., Adachi, H., Sobue, G., Breedlove, S. M., Lieberman, A. P., & Jordan, C. L. (2019). Disease Affects Bdnf Expression in Synaptic and Extrasynaptic Regions of Skeletal Muscle of Three SBMA Mouse Models. International Journal of Molecular Sciences, 20(6), 1314. https://doi.org/10.3390/ijms20061314