Therapeutic Targeting of Collective Invasion in Ovarian Cancer


Abstract
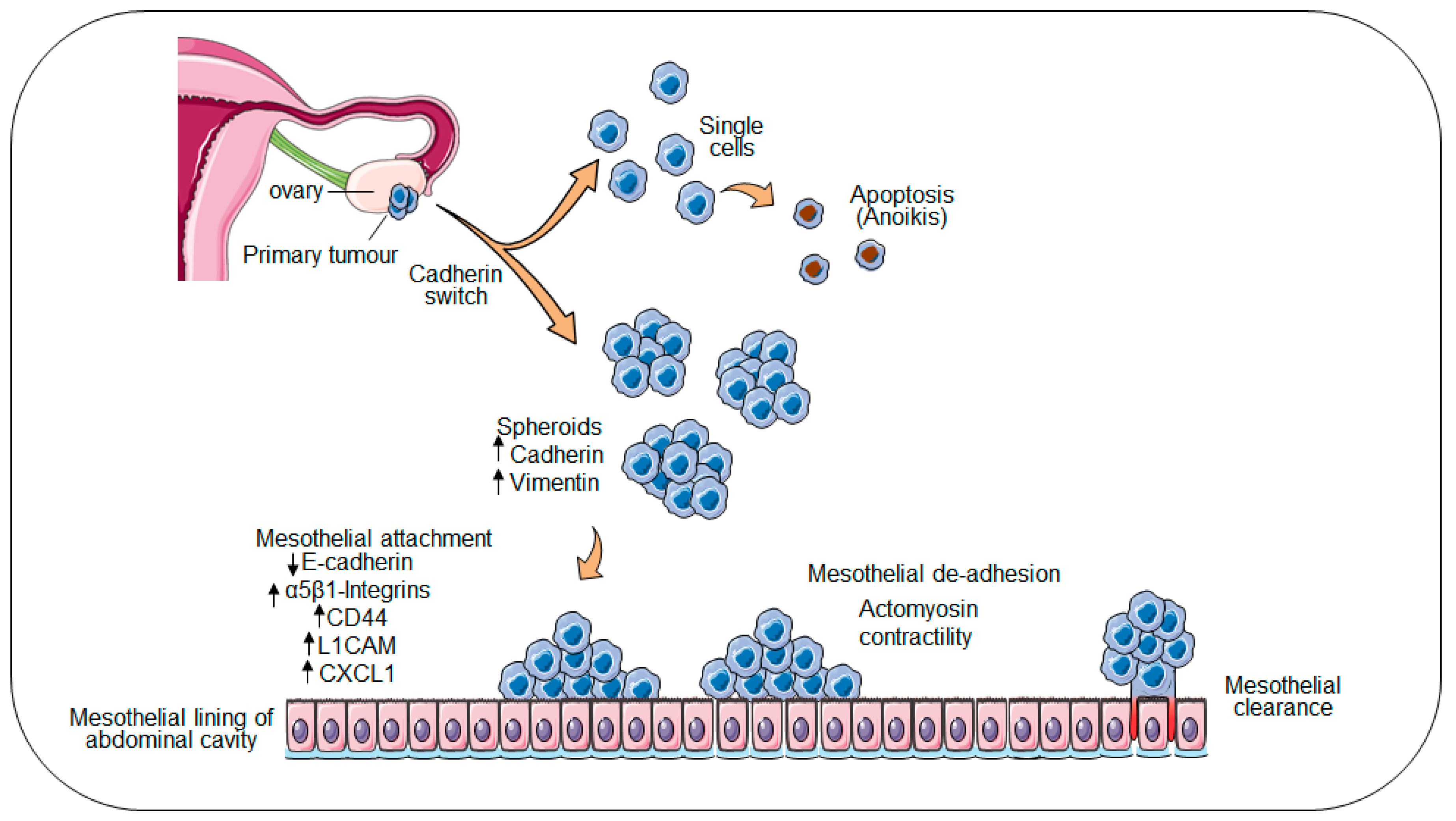
:1. Ovarian Cancer: A Unique Mode of Metastasis
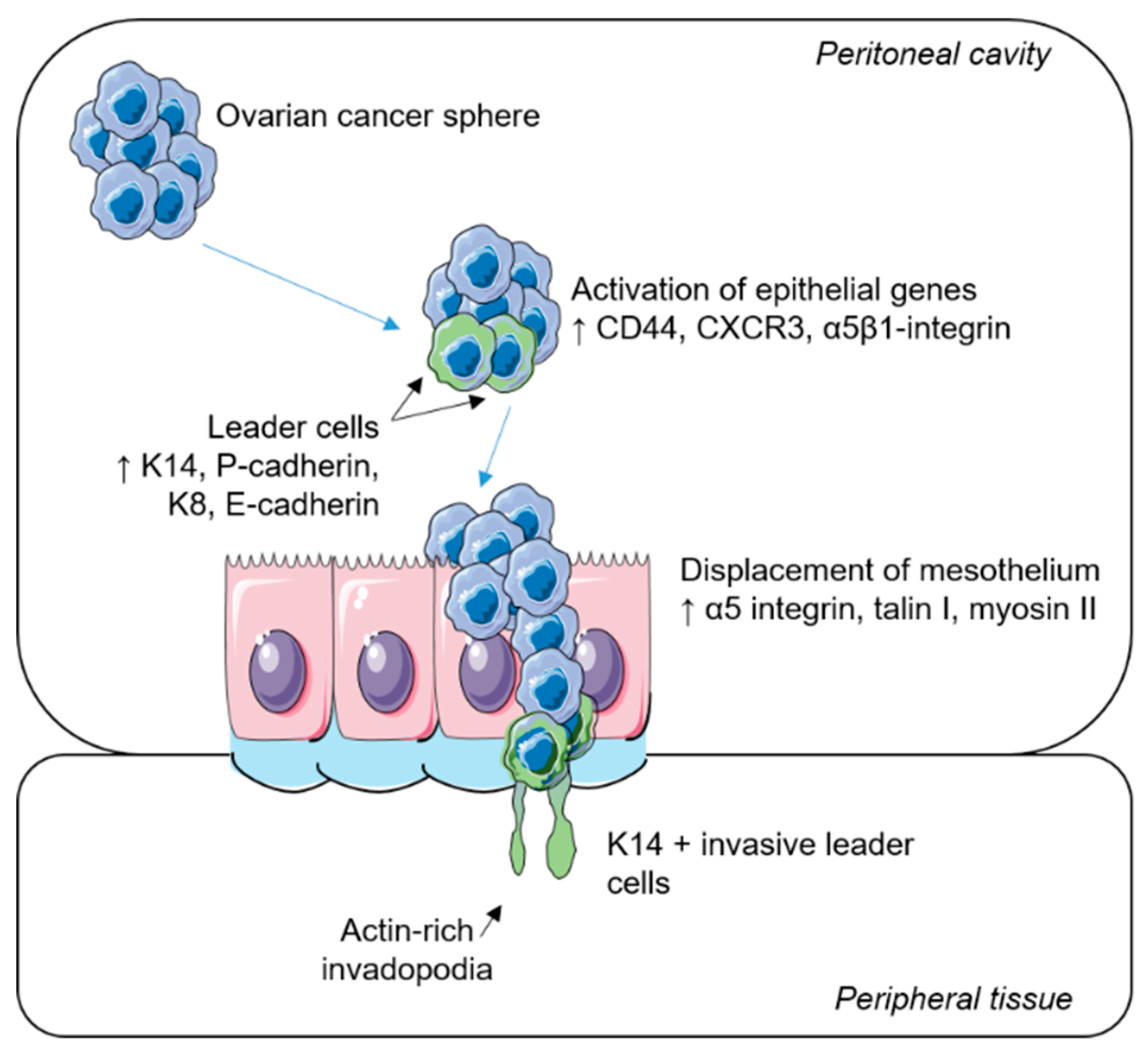
2. Collective Invasion and Leader Cells
3. Leader Cells and Progenitor-Like Properties
4. Targeting Leader Cells as a Novel Approach to Ovarian Cancer Management
5. Current Standard-of-Care in Ovarian Cancer Therapy
6. Targeting Leader Cell-Directed Processes: Collective Invasion and the Invasive Front
7. Strategies to Inhibit Collective Invasion in Ovarian Cancers
7.1. Cytoskeletal Stability
7.2. Rho Kinase Inhibition
7.3. Other Kinase Targets
8. Strategies to Disrupt Attachment and Invasion at the Invasive Interface
9. Molecular Targets in Leader Cells
Acknowledgments
Conflicts of Interest
References
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Kenny, H.A.; Krausz, T.; Yamada, S.D.; Lengyel, E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int. J. Cancer 2007, 121, 1463–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleson, K.M.; Hansen, L.K.; Skubitz, A.P. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin. Exp. Metastasis 2004, 21, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Boente, M.P.; Pambuccian, S.E.; Skubitz, A.P. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J. Transl. Med. 2006, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K. Getting to Know Ovarian Cancer Ascites: Opportunities for Targeted Therapy-Based Translational Research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef]
- Patel Ila, S.; Madan, P.; Getsios, S.; Bertrand Monique, A.; MacCalman, C.D. Cadherin switching in ovarian cancer progression. Int. J. Cancer 2003, 106, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053. [Google Scholar] [CrossRef]
- Elloul, S.; Silins, I.; Trope, C.G.; Benshushan, A.; Davidson, B.; Reich, R. Expression of E-cadherin transcriptional regulators in ovarian carcinoma. Virchows Arch 2006, 449, 520–528. [Google Scholar] [CrossRef]
- Aguilar-Gallardo, C.; Rutledge, E.C.; Martinez-Arroyo, A.M.; Hidalgo, J.J.; Domingo, S.; Simon, C. Overcoming challenges of ovarian cancer stem cells: Novel therapeutic approaches. Stem Cell Rev. 2012, 8, 994–1010. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef]
- Sodek, K.L.; Murphy, K.J.; Brown, T.J.; Ringuette, M.J. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev. 2012, 31, 397–414. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A.; Kansas, G.S.; Niloff, J.; DeFranzo, B.; Kim, Y.; Ottensmeier, C. Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by CD44H. Cancer Res. 1993, 53, 3830–3838. [Google Scholar] [PubMed]
- Strobel, T.; Swanson, L.; Cannistra, S.A. In vivo inhibition of CD44 limits intra-abdominal spread of a human ovarian cancer xenograft in nude mice: A novel role for CD44 in the process of peritoneal implantation. Cancer Res. 1997, 57, 1228–1232. [Google Scholar]
- Kenny, H.A.; Chiang, C.Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, R.C.; Burleson, K.M.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Ruff, L.E.; Skubitz, A.P. Beta 1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am. J. Pathol. 2001, 159, 2071–2080. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144. [Google Scholar] [CrossRef]
- Landen, C.N.; Kim, T.J.; Lin, Y.G.; Merritt, W.M.; Kamat, A.A.; Han, L.Y.; Spannuth, W.A.; Nick, A.M.; Jennnings, N.B.; Kinch, M.S.; et al. Tumor-selective response to antibody-mediated targeting of alphavbeta3 integrin in ovarian cancer. Neoplasia 2008, 10, 1259–1267. [Google Scholar] [CrossRef]
- Shield, K.; Riley, C.; Quinn, M.A.; Rice, G.E.; Ackland, M.L.; Ahmed, N. Alpha2beta1 integrin affects metastatic potential of ovarian carcinoma spheroids by supporting disaggregation and proteolysis. J. Carcinog. 2007, 6, 11. [Google Scholar] [CrossRef]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, A.; Schlich, S.; Issa, Y.; Gschwend, V.; Wenger, T.; Herr, I.; Marme, A.; Bourbie, S.; Altevogt, P.; Gutwein, P. L1 on ovarian carcinoma cells is a binding partner for Neuropilin-1 on mesothelial cells. Cancer Lett. 2006, 239, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.; Novak-Hofer, I.; Gast, D.; Gschwend, V.; Moldenhauer, G.; Gruenberg, J.; Honer, M.; Schubiger, P.; Altevogt, P.; Krueger, A. Efficient Inhibition of Intra-Peritoneal Tumor Growth and Dissemination of Human Ovarian Carcinoma Cells in Nude Mice by Anti-L1-Cell Adhesion Molecule Monoclonal Antibody Treatment. Cancer Res. 2006, 66, 936–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knogler, K.; Gruenberg, J.; Zimmermann, K.; Cohrs, S.; Honer, M.; Ametamey, S.; Altevogt, P.; Fogel, M.; Schubiger, P.; Novak-Hofer, I. Copper-67 Radioimmunotherapy and Growth Inhibition by Anti-L1-Cell Adhesion Molecule Monoclonal Antibodies in a Therapy Model of Ovarian Cancer Metastasis. Clin. Cancer Res. 2007, 13, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rooper, L.; Xie, J.; Kajdacsy-Balla, A.A.; Barbolina, M.V. Fractalkine receptor CX(3)CR1 is expressed in epithelial ovarian carcinoma cells and required for motility and adhesion to peritoneal mesothelial cells. Mol. Cancer Res. 2012, 10, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Montell, D.J. Morphogenetic Cell Movements: Diversity from Modular Mechanical Properties. Science 2008, 322, 1502–1505. [Google Scholar] [CrossRef]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Enomoto, A.; Asai, N.; Kato, T.; Takahashi, M. Collective invasion of cancer: Perspectives from pathology and development. Pathol. Int. 2016, 66, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Beerling, E.; Oosterom, I.; Voest, E.; Lolkema, M.; van Rheenen, J. Intravital characterization of tumor cell migration in pancreatic cancer. Intravital 2016, 5, e1261773. [Google Scholar] [CrossRef] [Green Version]
- Sonoshita, M.; Itatani, Y.; Kakizaki, F.; Sakimura, K.; Terashima, T.; Katsuyama, Y.; Sakai, Y.; Taketo, M.M. Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHOGEF protein TRIO. Cancer Discov. 2015, 5, 198–211. [Google Scholar] [CrossRef]
- Hesse, K.; Satzger, I.; Schacht, V.; Köther, B.; Hillen, U.; Klode, J.; Schaper, K.; Gutzmer, R. Characterisation of Prognosis and Invasion of Cutaneous Squamous Cell Carcinoma by Podoplanin and E-Cadherin Expression. Dermatology 2016, 232, 558–565. [Google Scholar] [CrossRef]
- Hegerfeldt, Y.; Tusch, M.; Brocker, E.B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar]
- Ewald, A.J.; Huebner, R.J.; Palsdottir, H.; Lee, J.K.; Perez, M.J.; Jorgens, D.M.; Tauscher, A.N.; Cheung, K.J.; Werb, Z.; Auer, M. Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci. 2012, 125 Pt 11, 2638–2654. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639. [Google Scholar] [CrossRef]
- Haney, S.; Konen, J.; Marcus, A.I.; Bazhenov, M. The complex ecosystem in non small cell lung cancer invasion. PLoS Comput. Biol. 2018, 14, e1006131. [Google Scholar] [CrossRef]
- Khalil, A.A.; Friedl, P. Determinants of leader cells in collective cell migration. Integr. Biol. 2010, 2, 568–574. [Google Scholar] [CrossRef]
- Volkmer, J.P.; Sahoo, D.; Chin, R.K.; Ho, P.L.; Tang, C.; Kurtova, A.V.; Willingham, S.B.; Pazhanisamy, S.K.; Contreras-Trujillo, H.; Storm, T.A.; et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc. Natl. Acad. Sci. USA 2012, 109, 2078–2083. [Google Scholar] [CrossRef] [Green Version]
- Venhuizen, J.-H.; Zegers, M.M. Making Heads or Tails of It: Cell–Cell Adhesion in Cellular and Supracellular Polarity in Collective Migration. Cold Spring Harbor Perspect. Biol. 2017, 9, a027854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small Gtpases 2015, 6, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Actomyosin contractility and collective migration: May the force be with you. Curr. Opin. Cell Biol. 2017, 48, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Witz, C.A.; Montoya-Rodriguez, I.A.; Cho, S.; Centonze, V.E.; Bonewald, L.F.; Schenken, R.S. Composition of the Extracellular Matrix of the Peritoneum. J. Soc. Gynecol. Investig. 2001, 8, 299–304. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Neri, S.; Ishii, G.; Hashimoto, H.; Kuwata, T.; Nagai, K.; Date, H.; Ochiai, A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int. J. Cancer 2015, 137, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- George, P.; Paraskevopoulou, V.; Vasilaki, E.; Kanaki, Z.; Paschalidis, N.; Klinakis, A. KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat. Commun. 2016, 7, 11914. [Google Scholar] [Green Version]
- Xiao-Lei, G.; Wu, J.S.; Cao, M.X.; Gao, S.Y.; Cen, X.; Jiang, Y.P.; Wang, S.S.; Tang, Y.J.; Chen, Q.M.; Liang, X.H.; et al. Cytokeratin-14 contributes to collective invasion of salivary adenoid cystic carcinoma. PLoS ONE 2017, 12, e0171341. [Google Scholar]
- Hu, W.-Y.; Hu, D.P.; Xie, L.; Li, Y.; Majumdar, S.; Nonn, L.; Hu, H.; Shioda, T.; Prins, G.S. solation and functional interrogation of adult human prostate epithelial stem cells at single cell resolution. Stem Cell Res. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Qian, W.; Zhang, H.; Liang, Y.; Wu, M.; Zhang, Y.; Zhang, X.; Gao, Q.; Li, Y. SOX2 Is a Marker for Stem-like Tumor Cells in Bladder Cancer. Stem Cell Rep. 2017, 9, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Barry, W.; Penson, R.T.; Konstantinopoulos, P.A.; Luo, W.; Hoffman, M.A.; Horowitz, N.S.; Farooq, S.; Dizon, D.S.; Stover, E.; et al. Phase II study of pembrolizumab (pembro) combined with pegylated liposomal doxorubicin (PLD) for recurrent platinum-resistant ovarian, fallopian tube or peritoneal cancer. Gynecol. Oncol. 2018, 149, 24. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer: Phase 1b Results From the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Aghajanian, C.; Finkler, N.J.; Rutherford, T.; Smith, D.A.; Yi, J.; Parmar, H.; Nycum, L.R.; Sovak, M.A. OCEANS: A randomized, double-blinded, placebo-controlled phase III trial of chemotherapy with or without bevacizumab (BEV) in patients with platinum-sensitive recurrent epithelial ovarian (EOC), primary peritoneal (PPC), or fallopian tube cancer (FTC). J. Clin. Oncol. 2011, 29 (Suppl. 18), LBA5007. [Google Scholar] [CrossRef]
- Morrison, J.; Thoma, C.; Goodall, R.J.; Lyons, T.J.; Gaitskell, K.; Wiggans, A.J.; Bryant, A. Epidermal growth factor receptor blockers for the treatment of ovarian cancer. Cochrane Database Syst. Rev. 2018, 10, Cd007927. [Google Scholar] [CrossRef]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27 (Suppl. 1), i33–i39. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.D.; Clamp, A.R.; Evans, D.G.R.; Edmondson, R.J.; Jayson, G.C. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother. Pharmacol. 2018, 81, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28. [Google Scholar]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia Are Required for Cancer Cell Extravasation and Are a Therapeutic Target for Metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Trendowski, M. Exploiting the cytoskeletal filaments of neoplastic cells to potentiate a novel therapeutic approach. Biochim. Biophys. Acta 2014, 1846, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Bousquet, P.F.; Paulsen, L.A.; Fondy, C.; Lipski, K.M.; Loucy, K.J.; Fondy, T.P. Effects of cytochalasin B in culture and in vivo on murine Madison 109 lung carcinoma and on B16 melanoma. Cancer Res. 1990, 50, 1431–1439. [Google Scholar]
- Senderowicz, A.M.; Kaur, G.; Sainz, E.; Laing, C.; Inman, W.D.; Rodriguez, J.; Crews, P.; Malspeis, L.; Grever, M.R.; Sausville, E.A.; et al. Jasplakinolide’s inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J. Natl. Cancer Inst. 1995, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Jalilian, I.; Heu, C.; Cheng, H.; Freittag, H.; Desouza, M.; Stehn, J.R.; Bryce, N.S.; Whan, R.M.; Hardeman, E.C.; Fath, T.; et al. Cell elasticity is regulated by the tropomyosin isoform composition of the actin cytoskeleton. PLoS ONE 2015, 10, e0126214. [Google Scholar] [CrossRef] [PubMed]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.B.B.; Sequeira, V.B.; Butler, T.L.; et al. A Novel Class of Anticancer Compounds Targets the Actin Cytoskeleton in Tumor Cells. Cancer Res. 2013, 73, 5169. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, M.S.; Ashley, S.W.; Whang, E.E. Inhibition of pancreatic adenocarcinoma cellular invasiveness by blebbistatin: A novel myosin II inhibitor. Biochem. Biophys. Res. Commun. 2004, 313, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.; Taleski, G.; Sontag, E. The protein serine/threonine phosphatases PP2A, PP1 and calcineurin: A triple threat in the regulation of the neuronal cytoskeleton. Mol. Cell. Neurosci. 2017, 84, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Gandalovicova, A.; Rosel, D.; Fernandes, M.; Vesely, P.; Heneberg, P.; Cermak, V.; Petruzelka, L.; Kumar, S.; Sanz-Moreno, V.; Brabek, J. Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Sadok, A.; Marshall, C.J. Rho GTPases: Masters of cell migration. Small GTPases 2014, 5, e29710. [Google Scholar] [CrossRef] [PubMed]
- Reffay, M.; Parrini, M.; Cochet-Escartin, O.; Ladoux, B.; Buguin, A.; Coscoy, S.; Amblard, F.; Camonis, J.; Silberzan, P. Interplay of RhoA and mechanical forces in collective cell migration driven by leader cells. Nat. Cell Biol. 2014, 16, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Gou, W.-F.; Xiu, Y.-L.; Zheng, H.-C.; Zong, Z.-H.; Takano, Y.; Zhao, Y. The involvement of RhoA and Wnt-5a in the tumorigenesis and progression of ovarian epithelial carcinoma. Int. J. Mol. Sci. 2013, 14, 24187–24199. [Google Scholar] [CrossRef]
- Horiuchi, A.; Kikuchi, N.; Osada, R.; Wang, C.; Hayashi, A.; Nikaido, T.; Konishi, I. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with Lovastatin may be useful for ovarian cancer. Cancer Sci. 2008, 99, 2532–2539. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, W.; Kang, J.; Liu, Q.; Nie, M. Knockdown of RhoA expression alters ovarian cancer biological behavior in vitro and in nude mice. Oncol. Rep. 2015, 34, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, L.; Shan, S.; Sun, Y.; Mao, Y. KRT14 promoting invasion and migration of lung cancer cells through ROCK-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 795–803. [Google Scholar]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Kümper, S.; Mardakheh, F.K.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jørgensen, C.; Guichard, S.; et al. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 2016, 5, e12203. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Nakao, H.; Yoshizaki, H.; Shiratsuchi, M.; Shigyo, H.; Yamada, H.; Ozawa, T.; Totsuka, J.; Hidaka, H. Development of specific Rho-kinase inhibitors and their clinical application. BBA-Proteins Proteom. 2005, 1754, 245–252. [Google Scholar] [CrossRef]
- Deng, L.; Li, G.; Li, R.; Liu, Q.; He, Q.; Zhang, J. Rho-kinase inhibitor, fasudil, suppresses glioblastoma cell line progression in vitro and in vivo. Cancer Biol. Ther. 2010, 9, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, Y.; Wang, S.; Shi, W. Effect of fasudil on growth, adhesion, invasion, and migration of 95D lung carcinoma cells in vitro. Can. J. Physiol. Pharmacol. 2010, 88, 874–879. [Google Scholar] [CrossRef]
- Yang, X.; Di, J.; Zhang, Y.; Zhang, S.; Lu, J.; Liu, J.; Shi, W. The Rho-kinase inhibitor inhibits proliferation and metastasis of small cell lung cancer. Biomed. Pharmacother. 2012, 66, 221–227. [Google Scholar] [CrossRef]
- Hu, K.; Wang, Z.; Tao, Y. [Suppression of hepatocellular carcinoma invasion and metastasis by Rho-kinase inhibitor Fasudil through inhibition of BTBD7-ROCK2 signaling pathway]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014, 39, 1221–1227. [Google Scholar]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; del Monte-Nieto, G.; Conway, J.R.W.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef] [Green Version]
- Ogata, S.; Morishige, K.I.; Sawada, K.; Hashimoto, K.; Mabuchi, S.; Kawase, C.; Ooyagi, C.; Sakata, M.; Kimura, T. Fasudil Inhibits Lysophosphatidic Acid-Induced Invasiveness of Human Ovarian Cancer Cells. Int. J. Gynecol. Cancer 2009, 19, 1473–1480. [Google Scholar] [CrossRef]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279. [Google Scholar] [CrossRef]
- Ohta, T.; Takahashi, T.; Shibuya, T.; Amita, M.; Henmi, N.; Takahashi, K.; Kurachi, H. Inhibition of the Rho/ROCK pathway enhances the efficacy of cisplatin through the blockage of hypoxia-inducible factor-1alpha in human ovarian cancer cells. Cancer Biol. 2012, 13, 25–33. [Google Scholar] [CrossRef]
- Yap, T.A.; Walton, M.I.; Grimshaw, K.M.; Poele, R.H.t.; Eve, P.D.; Valenti, M.R.; Brandon, A.K.d.; Martins, V.; Zetterlund, A.; Heaton, S.P.; et al. AT13148 Is a Novel, Oral Multi-AGC Kinase Inhibitor with Potent Pharmacodynamic and Antitumor Activity. Clin. Cancer Res. 2012, 18, 3912. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Kumar, R.; Yap, T.A.; Ruddle, R.; Decordova, S.; Jones, P.; Halbert, G.; Garrett, M.D.; McLeod, R.; Backholer, Z.; et al. A first-in-human study of the dual ROCK I/II inhibitor, AT13148, in patients with advanced cancers. J. Clin. Oncol. 2015, 33 (Suppl. 15), 2566. [Google Scholar] [CrossRef]
- Salhia, B.; Rutten, F.; Nakada, M.; Beaudry, C.; Berens, M.; Kwan, A.; Rutka, J.T. Inhibition of Rho-Kinase Affects Astrocytoma Morphology, Motility, and Invasion through Activation of Rac1. Cancer Res. 2005, 65, 8792. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Yashiro, M.; Kato, Y.; Shinto, O.; Kashiwagi, S.; Hirakawa, K. RhoA/ROCK signaling mediates plasticity of scirrhous gastric carcinoma motility. Clin. Exp. Metastasis 2011, 28, 627–636. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef] [Green Version]
- Mayer, E.L.; Krop, I.E. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin. Cancer Res. 2010, 16, 3526–3532. [Google Scholar] [CrossRef]
- Wiener, J.R.; Windham, T.C.; Estrella, V.C.; Parikh, N.U.; Thall, P.F.; Deavers, M.T.; Bast, R.C.; Mills, G.B.; Gallick, G.E. Activated SRC protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol. Oncol. 2003, 88, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Yang, J. Targeting invadopodia to block breast cancer metastasis. Oncotarget 2011, 2, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.O.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR–Src–Arg–Cortactin Pathway Mediates Functional Maturation of Invadopodia and Breast Cancer Cell Invasion. Cancer Res. 2011, 71, 1730. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Schilder, R.J.; Brady, W.E.; Lankes, H.A.; Fiorica, J.V.; Shahin, M.S.; Zhou, X.C.; Mannel, R.S.; Pathak, H.B.; Hu, W.; Alpaugh, R.K.; et al. Phase II evaluation of dasatinib in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 127, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.B.; Smith, D.C.; Berger, R.; Kurzrock, R.; Vogelzang, N.J.; Sella, A.; Wheler, J.; Lee, Y.; Foster, P.G.; Weitzman, R.; et al. A phase 2 randomised discontinuation trial of cabozantinib in patients with ovarian carcinoma. Eur. J. Cancer 2017, 83, 229–236. [Google Scholar] [CrossRef]
- Chekerov, R.; Hilpert, F.; Mahner, S.; El-Balat, A.; Harter, P.; de Gregorio, N.; Fridrich, C.; Markmann, S.; Potenberg, J.; Lorenz, R.; et al. Sorafenib plus topotecan versus placebo plus topotecan for platinum-resistant ovarian cancer (TRIAS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 1247–1258. [Google Scholar] [CrossRef]
- Schoffski, P.; Gordon, M.; Smith, D.C.; Kurzrock, R.; Daud, A.; Vogelzang, N.J.; Lee, Y.; Scheffold, C.; Shapiro, G.I. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur. J. Cancer 2017, 86, 296–304. [Google Scholar] [CrossRef]
- Matei, D.; Sill, M.W.; Lankes, H.A.; DeGeest, K.; Bristow, R.E.; Mutch, D.; Yamada, S.D.; Cohn, D.; Calvert, V.; Farley, J.; et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: A gynecologic oncology group trial. J. Clin. Oncol. 2011, 29, 69–75. [Google Scholar] [CrossRef]
- Ramasubbaiah, R.; Perkins, S.M.; Schilder, J.; Whalen, C.; Johnson, C.S.; Callahan, M.; Jones, T.; Sutton, G.; Matei, D. Sorafenib in combination with weekly topotecan in recurrent ovarian cancer, a phase I/II study of the Hoosier Oncology Group. Gynecol. Oncol. 2011, 123, 499–504. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Thompson, D.S.; Bismayer, J.A.; Gian, V.G.; Merritt, W.M.; Whorf, R.C.; Finney, L.H.; Dudley, B.S. Paclitaxel/carboplatin with or without sorafenib in the first-line treatment of patients with stage III/IV epithelial ovarian cancer: A randomized phase II study of the Sarah Cannon Research Institute. Cancer Med. 2015, 4, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.; Liu, Y.; Wang, B.; Li, R.; Sebti, S.M. Identification of novel ROCK inhibitors with anti-migratory and anti-invasive activities. Oncogene 2013, 33, 550. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NCT00585052. A Phase II Study of Interaction of Lovastatin and Paclitaxel For Patients with Refractory or Relapsed Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00585052 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT02943317. Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Preliminary Clinical Activity of Defactinib in Combination with Avelumab in Epithelial Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02943317 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT00671788. A Phase II Evaluation of Dasatinib (Sprycel®, NSC #732517) in the Treatment of Persistent or Recurrent Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00671788 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT00940225. Study of Cabozantinib (XL184) in Adults with Advanced Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT00940225 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT01716715. Cabozantinib or Paclitaxel in Treating Patients with Persistent or Recurrent Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cavity Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01716715 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT00093626. Sorafenib in Treating Patients with Persistent or Recurrent Ovarian Epithelial or Peritoneal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00093626 (accessed on 13 December 2018).
- ClinicalTrials.gov. NCT00390611. Paclitaxel and Carboplatin with or without Sorafenib in the First-Line Treatment Of Patients With Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00390611 (accessed on 13 December 2018).
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase II, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. NCT00516841: A Phase 2, Single-Arm Study of Volociximab Monotherapy in Subjects with Platinum-Resistant Advanced Epithelial Ovarian Cancer or Primary Peritoneal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00516841 (accessed on 25 February 2019).
- ClinicalTrials.gov. NCT01670799. Availability & Effect of Post-OP Ketorolac on Ovarian, Fallopian Tube or Primary Peritoneal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01670799 (accessed on 13 December 2018).
- Guo, Y.; Kenney, S.R.; Cook, L.S.; Adams, S.F.; Rutledge, T.; Romero, E.; Oprea, T.; Sklar, L.A.; Bedrick, E.; Wiggins, C.L.; et al. A novel pharmacologic activity of ketorolac for therapeutic benefit in ovarian cancer patients. Clin. Cancer Res. 2015, 21, 5064–5072. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. NCT02470299. Evaluation of GTPase Inhibition by Post-Operative Intravenous Ketorolac in Ovarian Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02470299 (accessed on 13 December 2018).
- Yurchenco, P.D. Basement Membranes: Cell Scaffoldings and Signaling Platforms. Cold Spring Harbor Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef]
- Ricart, A.D.; Tolcher, A.W.; Liu, G.; Holen, K.; Schwartz, G.; Albertini, M.; Weiss, G.; Yazji, S.; Ng, C.; Wilding, G. Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: A phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 2008, 14, 7924–7929. [Google Scholar] [CrossRef]
- Chen, Q.; Manning, C.; Millar, H.; McCabe, F.; Ferrante, C.; Sharp, C.; Shahied-Arruda, L.; Doshi, P.; Nakada, M.; Anderson, G. CNTO 95, a fully human anti αv integrin antibody, inhibits cell signaling, migration, invasion, and spontaneous metastasis of human breast cancer cells. Off. J. Metastasis Res. Soc. 2008, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- O’Day, S.; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Thompson, J.A.; Gonzalez, R.; Trefzer, U.; et al. A randomised, phase II study of intetumumab, an anti-αv-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or—Dacarbazine in patients with stage IV metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sawada, K.; Kimura, T. Potential of Integrin Inhibitors for Treating Ovarian Cancer: A Literature Review. Cancers 2017, 9, 83. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.B.; Eggert, A.A.O.; Blass, K.; Bröcker, E.-B.; Becker, J.C. Expression of Matrix Metalloproteinases in the Microenvironment of Spontaneous and Experimental Melanoma Metastases Reflects the Requirements for Tumor Formation. Cancer Res. 2003, 63, 8221. [Google Scholar] [PubMed]
- Poincloux, R.; Lizárraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122, 3015. [Google Scholar] [CrossRef] [PubMed]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective Inhibition of Matrix Metalloproteinase-14 Blocks Tumor Growth, Invasion, and Angiogenesis. Cancer Res. 2009, 69, 1517. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, F.; Zhang, S.; Lu, J. Loss of RhoA expression prevents proliferation and metastasis of SPCA1 lung cancer cells in vitro. Biomed. Pharmacother. 2015, 69, 361–366. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Mizutani, T.; Kawabata, K.; Haga, H. Leader cells regulate collective cell migration via Rac activation in the downstream signaling of integrin β1 and PI3K. Sci. Rep. 2015, 5, 7656. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Target | Clinical Trial ID | Phase | Outcome Measures | Current Status | Refs |
|---|---|---|---|---|---|---|
| Lovastatin | RhoA | NCT00585052 | II | Tumour response rate in combination with paclitaxel for patients with relapsed ovarain cancer | Terminated due to slow accrual. | [113] |
| AT13148 | Multi-AGC kinase | NCT01585701 | I | Determine dosing and adverse events. Evaluate any response in patients with advanced-stage solid tumours including prostate, breast, and ovarian. | Completed. Preliminary data indicate tolerable on target effects. | [96,114] |
| Dasatinib | Src kinase | NCT00671788 | II | Progression Free Survivial at 6 months and tumour response in persistent or recurrent epithelial ovarian cancer using dasatinib as a monotherapy | Completed. dasatinib has minimal activity as a single agent in ovarian cancer (PFS 2.1 months). | [105,115] |
| Cabozantinib | Multi-kinases | NCT00940225 | II | Evaluate overall response rate and PFS in patients with advanced malignancies including melanoma, breast and ovarian cancer | Completed. Ovarian cancer patients showed the highest overall response rate (21.7%) and disease control rate was 50%. Platinum-sensitive patients achieved a longer PFS (6.9 months) than platinum-resistant patients (2.8 months). | [106,108,116] |
| NCT01716715 | II | Compare PFS in patients with persistent or recurrent ovarian cancer patients receiving cabozantinib or paclitaxel | Ongoing. | [117] | ||
| Sorafenib | NCT00093626 | II | Assess adverse events and PFS time in patients with persistent or recurrent ovarian cancer | Completed. Significant toxicity as a monotherapy with modest anti-tumour effect (PFS 2.1 months). | [109,118] | |
| NCT00526799 | I/II | Tolerability (Phase I) and response rate (Phase II) to treatment with sorafenib in combination with topotecan in patients with platinum-resistant or refractory-recurrent ovarian cancer | Terminated. Significant toxicity caused by sub-optimal doses of combination therapy associated with minimal clinical efficacy. | [110,119] | ||
| NCT00390611 | II | PFS over 2 years in patients with late-stage ovarian cancer receiving sorafenib in first-line treatment | Completed. Combination paclitaxel/carboplatin or paclitaxel/carboplatin/sorafenib had similar response rates and PFS (15.4 vs 16.3 months). The addition of sorafenib in first-line treatment caused increased toxicity. | [111,119] | ||
| Volociximab | α5β1-integrin | NCT00516841 | II | Evaluate efficacy of volociximab monotherapy by objective response rate and tumour response in patients with platinum resistant EOC | Terminated due to insufficient clinical activity. Volociximab was well tolerated; however, there were no complete or partial responses. | [120,121] |
| Ketorolac | Rac1/Cdc42 | NCT01670799 | 0 (Pilot) | Determine measurable R- and S-ketorolac in post-operative treated patients following cytoreductive ovarian cancer surgery | Ongoing. | [122] |
| NCT02470299 | I | Confirmation of drug specificity. Evaluation of overall survival and PFS in post-operative IV ketorolac treated ovarian cancer patients | Recruiting/ongoing. Preliminary data shows specific Rac1 and Cdc42 inhibition and potential prolonged survival in women receiving ketorolac. | [123,124] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moffitt, L.; Karimnia, N.; Stephens, A.; Bilandzic, M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 1466. https://doi.org/10.3390/ijms20061466
Moffitt L, Karimnia N, Stephens A, Bilandzic M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. International Journal of Molecular Sciences. 2019; 20(6):1466. https://doi.org/10.3390/ijms20061466
Chicago/Turabian StyleMoffitt, Laura, Nazanin Karimnia, Andrew Stephens, and Maree Bilandzic. 2019. "Therapeutic Targeting of Collective Invasion in Ovarian Cancer" International Journal of Molecular Sciences 20, no. 6: 1466. https://doi.org/10.3390/ijms20061466
APA StyleMoffitt, L., Karimnia, N., Stephens, A., & Bilandzic, M. (2019). Therapeutic Targeting of Collective Invasion in Ovarian Cancer. International Journal of Molecular Sciences, 20(6), 1466. https://doi.org/10.3390/ijms20061466