Interferon-Stimulated Genes—Mediators of the Innate Immune Response during Canine Distemper Virus Infection


Abstract
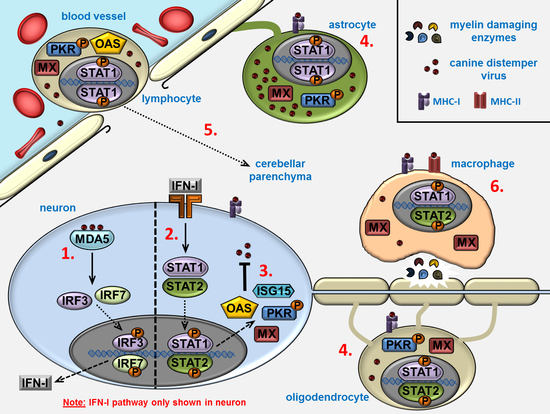
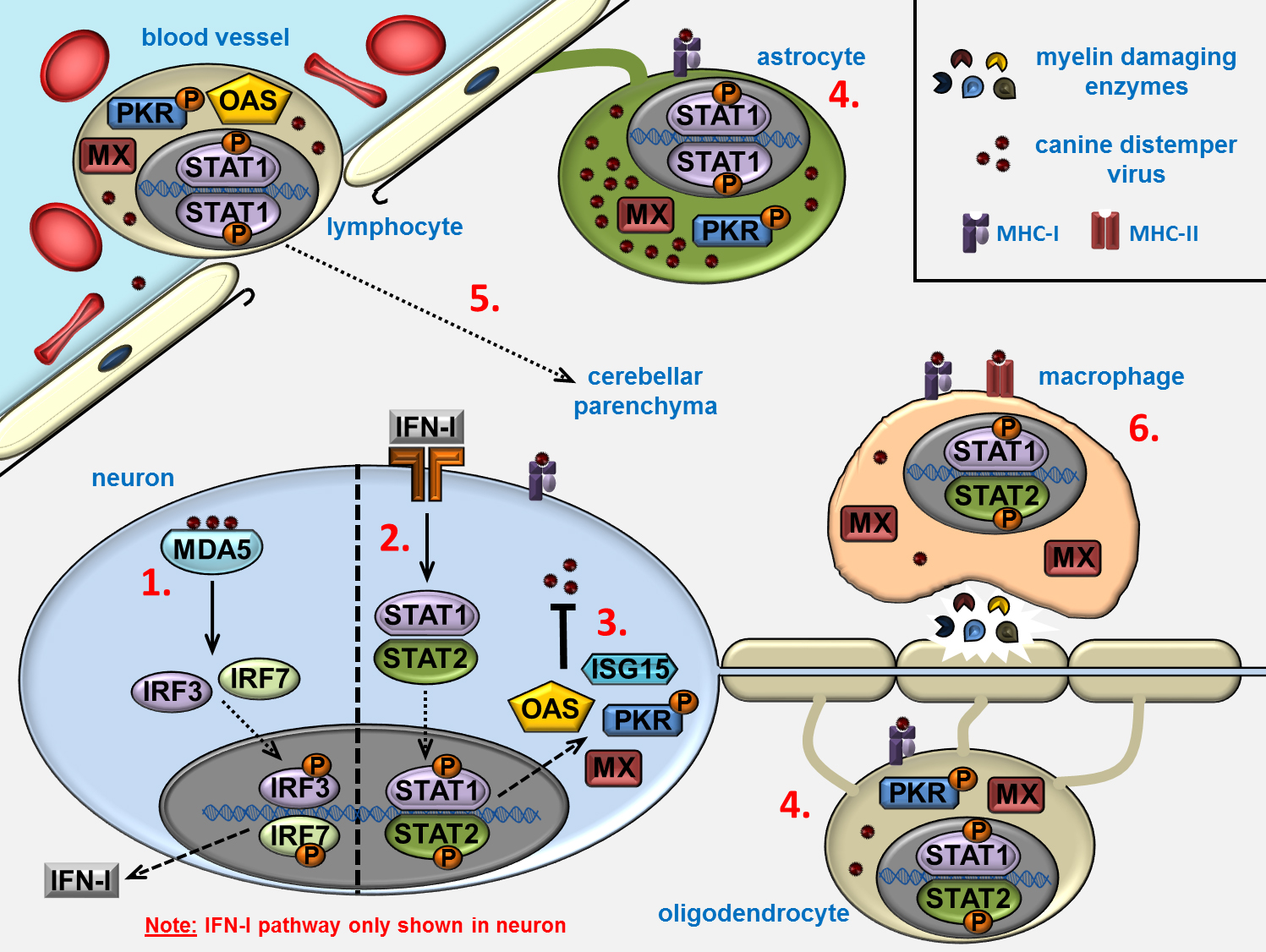
:
1. Introduction
2. Results
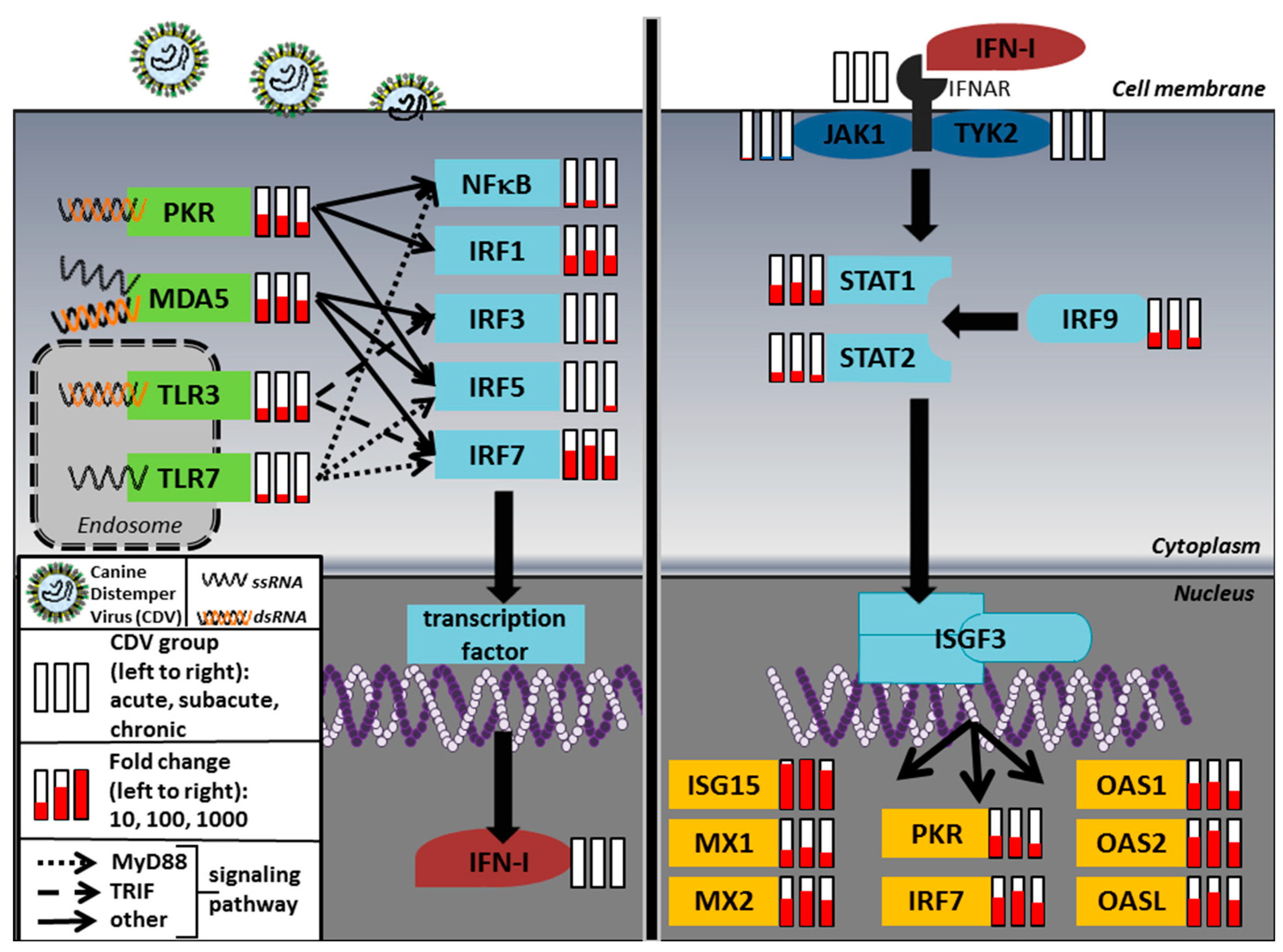
2.1. Microarray
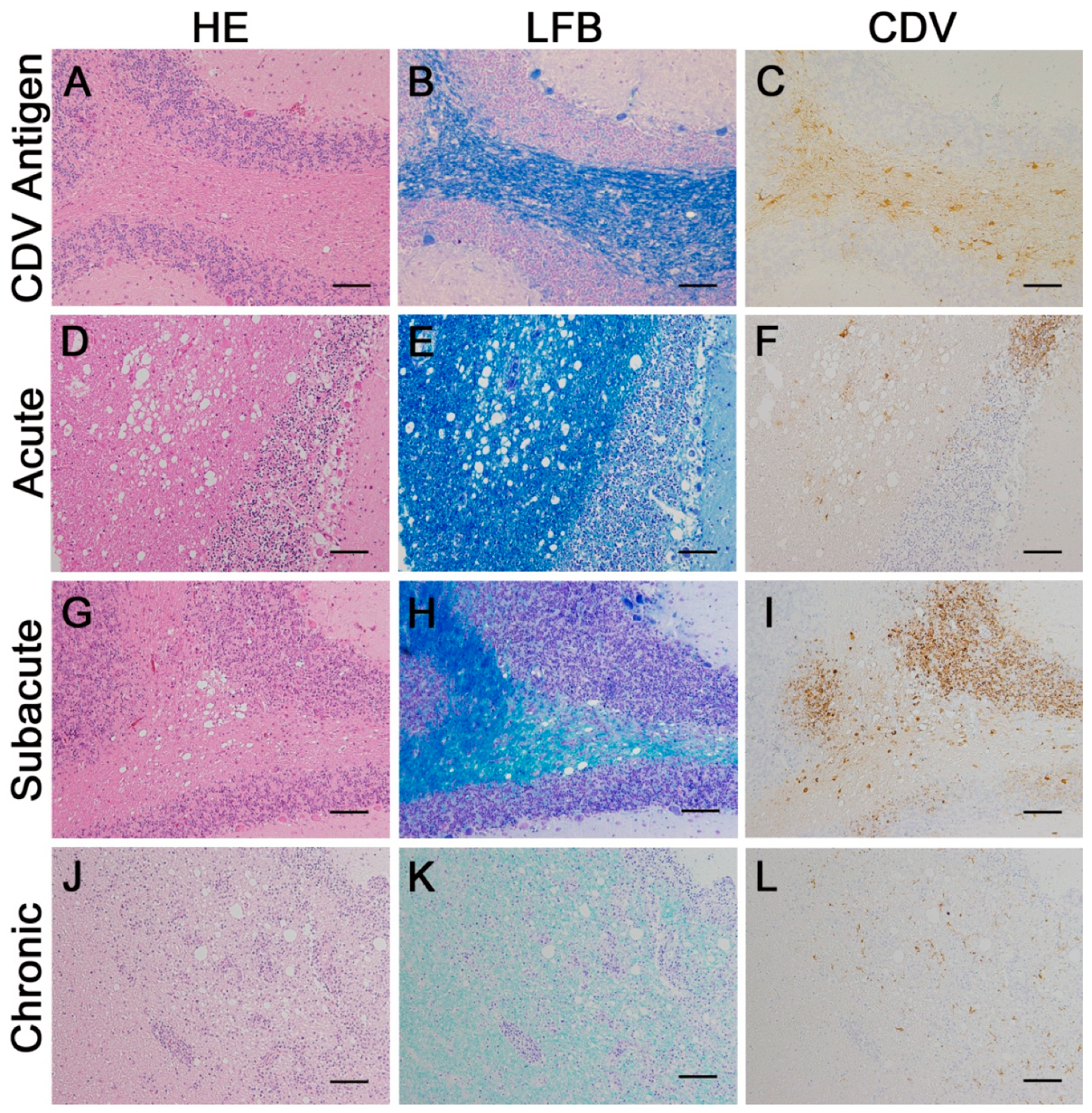
2.2. Classification of Histological Lesions
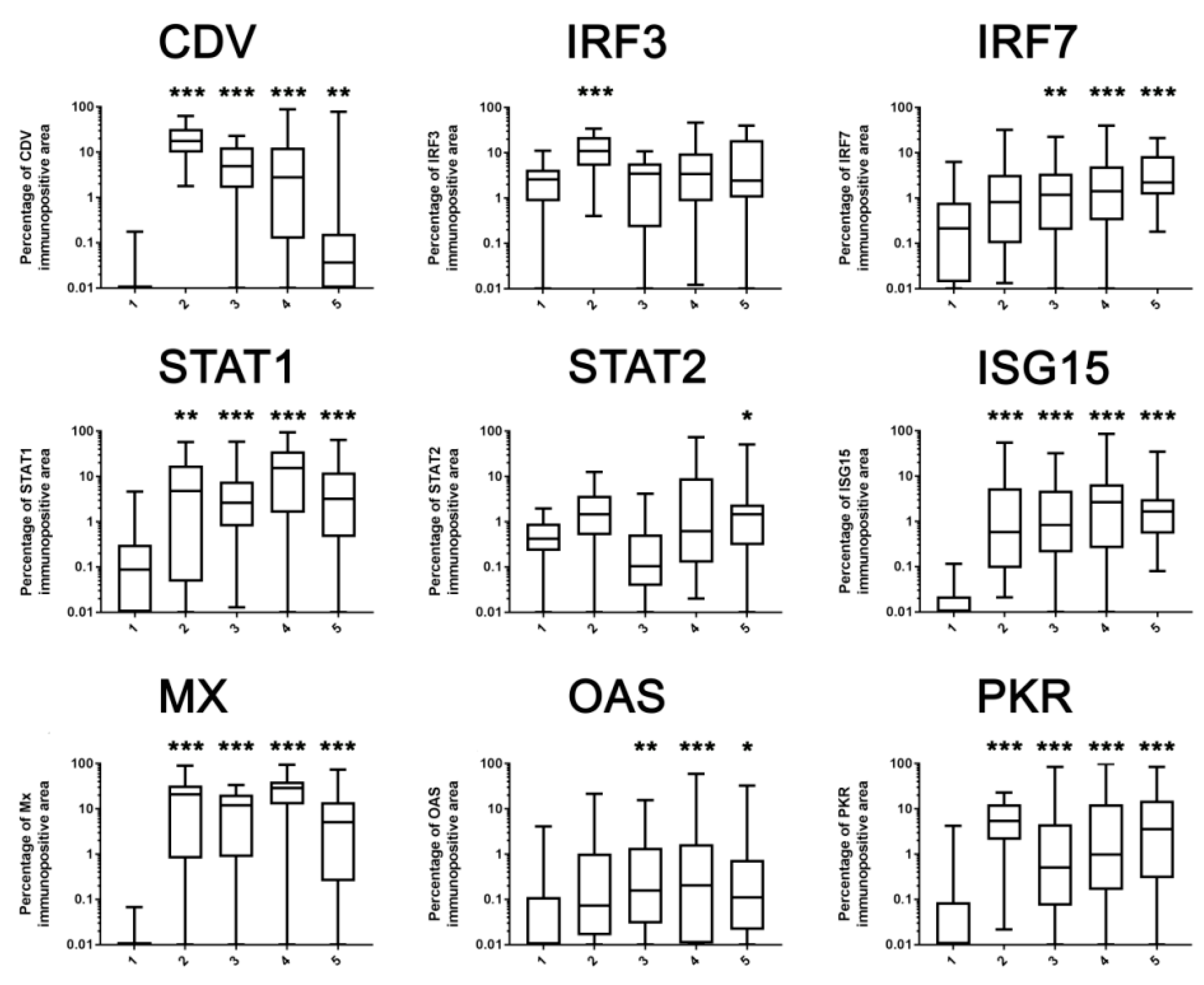
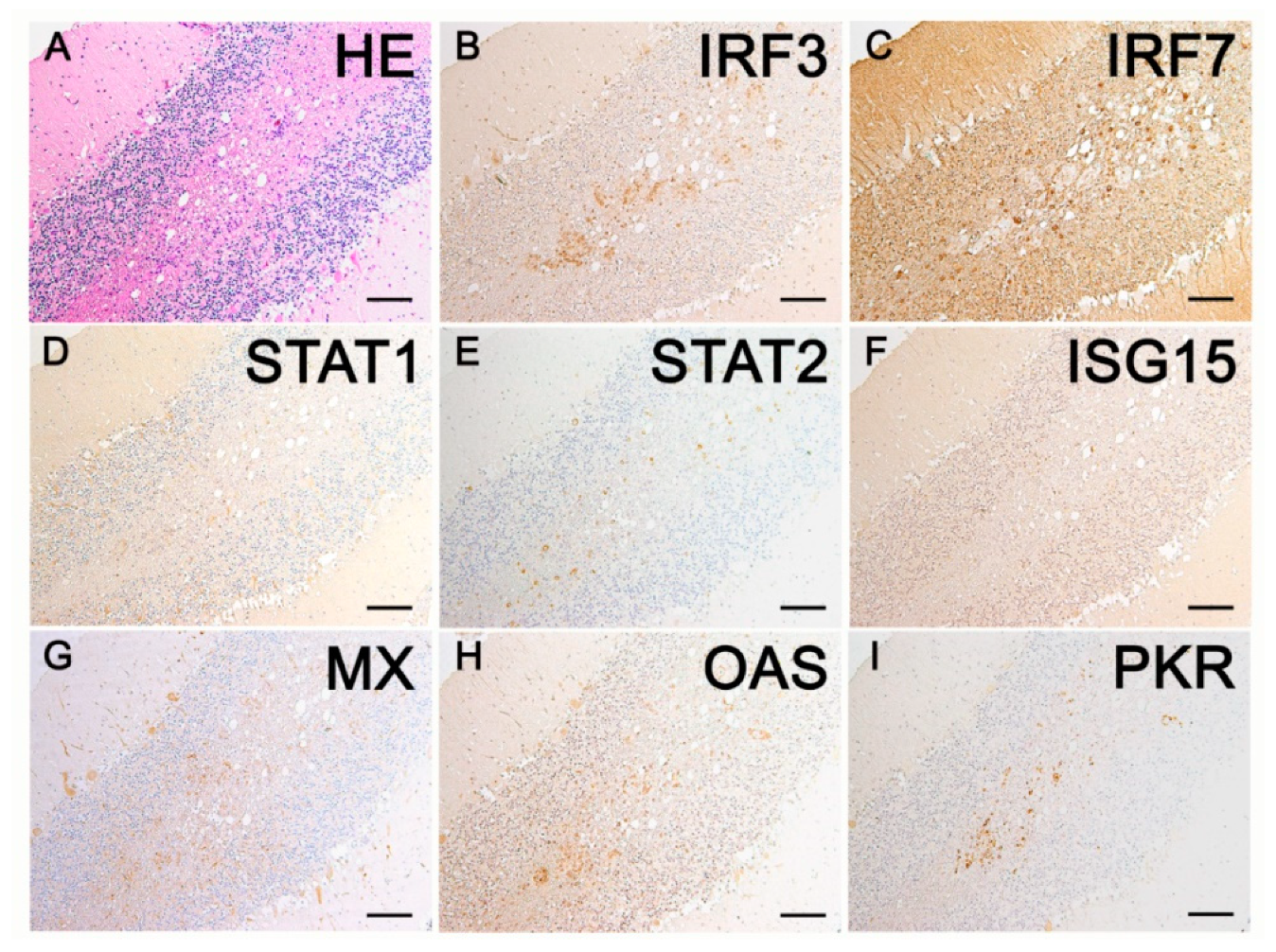
2.3. Immunohistochemistry
2.4. Correlation Analysis
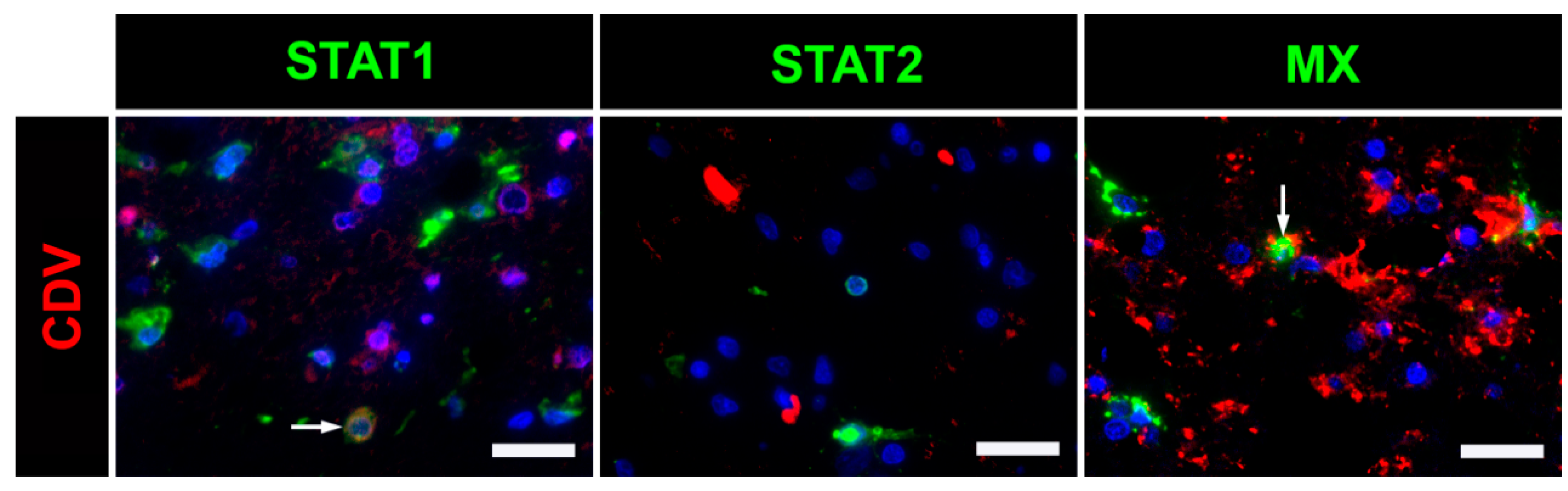
2.5. Immunofluorescence
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Histology and Classification of Cerebellar Lesions
4.3. Transcriptional Analysis of Interferon-stimulated Genes
4.4. Immunohistochemistry
4.5. Immunofluorescence Double-Labeling
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellini, W.J.; Englund, G.; Richardson, C.D.; Rozenblatt, S.; Lazzarini, R.A. Matrix genes of measles virus and canine distemper virus: Cloning, nucleotide sequences, and deduced amino acid sequences. J. Virol. 1986, 58, 408–416. [Google Scholar]
- Lempp, C.; Spitzbarth, I.; Puff, C.; Cana, A.; Kegler, K.; Techangamsuwan, S.; Baumgärtner, W.; Seehusen, F. New aspects of the pathogenesis of canine distemper leukoencephalitis. Viruses 2014, 6, 2571–2601. [Google Scholar] [CrossRef] [PubMed]
- Vandevelde, M.; Zurbriggen, A. Demyelination in canine distemper virus infection: A review. Acta Neuropathol. 2005, 109, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Beineke, A.; Puff, C.; Seehusen, F.; Baumgärtner, W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet. Immunol. Immunopathol. 2009, 127, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Baumgärtner, W. Virale Infektionen von Welpen und Junghunden unter Berücksichtigung der Staupevirusinfektion. Praktischer Tierarzt 1993, 47, 26–32. [Google Scholar]
- Nesseler, A.; Baumgärtner, W.; Gaedke, K.; Zurbriggen, A. Abundant expression of viral nucleoprotein mRNA and restricted translation of the corresponding viral protein in inclusion body polioencephalitis of canine distemper. J. Comp. Pathol. 1997, 116, 291–301. [Google Scholar] [CrossRef]
- Nesseler, A.; Baumgärtner, W.; Zurbriggen, A.; Örvell, C. Restricted virus protein translation in canine distemper virus inclusion body polioencephalitis. Vet. Microbiol. 1999, 69, 23–28. [Google Scholar] [CrossRef]
- Vandevelde, M.; Higgins, R.J.; Kristensen, B.; Kristensen, F.; Steck, A.J.; Kihm, U. Demyelination in experimental canine distemper virus infection: Immunological, pathologic, and immunohistological studies. Acta Neuropathol. 1982, 56, 285–293. [Google Scholar] [CrossRef]
- Baumgärtner, W.; Alldinger, S. The Pathogenesis of Canine Distemper Virus Induced Demyelination. In Experimental Models of Multiple Sclerosis; Lavi, E., Constantinescu, C.S., Eds.; Springer: New York, NY, USA, 2005; pp. 871–887. [Google Scholar]
- Summers, B.A.; Appel, M.J. Aspects of canine distemper virus and measles virus encephalomyelitis. Neuropathol. Appl. Neurobiol. 1994, 20, 525–534. [Google Scholar] [CrossRef]
- Renauld, J.C. Class II cytokine receptors and their ligands: Key antiviral and inflammatory modulators. Nat. Rev. Immunol. 2003, 3, 667–676. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biron, C.A. Role of early cytokines, including alpha and beta interferons (IFN-alpha/beta), in innate and adaptive immune responses to viral infections. Semin. Immunol. 1998, 10, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Klotz, D.; Baumgärtner, W.; Gerhauser, I. Type I interferons in the pathogenesis and treatment of canine diseases. Vet. Immunol. Immunopathol. 2017, 191, 80–93. [Google Scholar] [CrossRef]
- Gamero, A.M.; Hodge, D.L.; Reynolds, D.M.; Rodriguez-Galan, M.C.; Mohamadzadeh, M.; Young, H.A. Interferon-γ: Gene and Protein Structure, Transcription Regulation, and Actions. In The Interferons: Characterization and Application; Meager, A., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 85–111. [Google Scholar]
- Donnelly, R.P.; Kotenko, S.V. Interferon-lambda: A new addition to an old family. J. Interferon Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Xu, L.; Ren, L.; Qu, H.; Li, J.; Liang, J.; Liu, W.; Yang, L.; Luo, T. Functional characterization of canine interferon-lambda. J. Interferon Cytokine Res. 2014, 34, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, T.; Asano, A.; Usui, T.; Takeuchi, T.; Watanabe, Y.; Yamano, Y. Antiviral and antiproliferative effects of canine interferon-lambda1. Vet. Immunol. Immunopathol. 2013, 156, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Yang, L.; Wei, J.; He, S. Integrative genomic analyses on interferon-lambdas and their roles in cancer prediction. Int. J. Mol. Med. 2010, 25, 299–304. [Google Scholar] [PubMed]
- Durelli, L.; Verdun, E.; Barbero, P.; Bergui, M.; Versino, E.; Ghezzi, A.; Montanari, E.; Zaffaroni, M.; Independent Comparison of Interferon Trial Study, G. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: Results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet 2002, 359, 1453–1460. [Google Scholar] [CrossRef]
- Townsell, M.Y.; Pohlman, L.M.; Harkin, K.R. Pathology in practice. Canine distemper virus disease in a dog. J. Am. Vet. Med. Assoc. 2015, 246, 613–615. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.F. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccin. Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.J.; Gale, M., Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 2011, 1, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boo, K.H.; Yang, J.S. Intrinsic cellular defenses against virus infection by antiviral type I interferon. Yonsei Med. J. 2010, 51, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Pestka, S. The interferon receptors. Semin. Oncol. 1997, 24, S9–S18. [Google Scholar]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Reich, N.; Darnell, J.E., Jr. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 1988, 2, 383–393. [Google Scholar] [CrossRef]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, W.; Wang, X.; Zhang, X.; Tian, H.; Deng, H.; Zhang, L.; Gao, G. Identification of new type I interferon-stimulated genes and investigation of their involvement in IFN-beta activation. Protein Cell 2018, 9, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.R.; Abraham, R.; Sundaram, S.; Sreekumar, E. Interferon regulated gene (IRG) expression-signature in a mouse model of chikungunya virus neurovirulence. J. Neurovirol. 2017, 23, 886–902. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef]
- Kimoto, T. In vitro and in vivo properties of the virus causing natural canine distemper encephalitis. J. Gen. Virol. 1986, 67 Pt 3, 487–503. [Google Scholar] [CrossRef]
- Tsai, S.C.; Summers, B.A.; Appel, M.J. Interferon in cerebrospinal fluid. A marker for viral persistence of canine distemper encephalomyelitis. Arch. Virol. 1982, 72, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.F.; Ambrus, A.; Storts, R.W. Immunohistochemical evaluation of mx protein expression in canine encephalitides. Vet. Pathol. 2006, 43, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Svitek, N.; Gerhauser, I.; Goncalves, C.; Grabski, E.; Döring, M.; Kalinke, U.; Anderson, D.E.; Cattaneo, R.; von Messling, V. Morbillivirus control of the interferon response: Relevance of STAT2 and mda5 but not STAT1 for canine distemper virus virulence in ferrets. J. Virol. 2014, 88, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Yamamoto, A.; Fujino, M.; Sato, I.; Hosokawa-Kanai, T.; Tuchiya, K.; Ishihama, A.; Sokawa, Y. High level activity of 2’, 5’-oligoadenylate synthetase in dog serum. J. Vet. Med. Sci. 2004, 66, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Puff, C.; Wewetzer, K.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W. Transcriptional changes in canine distemper virus-induced demyelinating leukoencephalitis favor a biphasic mode of demyelination. PLoS ONE 2014, 9, e95917. [Google Scholar] [CrossRef]
- Von Recum-Knepper, J.; Sadewasser, A.; Weinheimer, V.K.; Wolff, T. Fluorescence-Activated Cell Sorting-Based Analysis Reveals an Asymmetric Induction of Interferon-Stimulated Genes in Response to Seasonal Influenza A Virus. J. Virol. 2015, 89, 6982–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xin, X.; Wang, M.; Han, L.; Li, J.; Hao, Y.; Zheng, C.; Shen, C. Myxovirus resistance protein A inhibits hepatitis C virus replication through JAK-STAT pathway activation. Arch. Virol. 2018, 163, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Vig, P.J.S.; Lu, D.; Paul, A.M.; Kuwar, R.; Lopez, M.; Stokic, D.S.; Leis, A.A.; Garrett, M.R.; Bai, F. Differential Expression of Genes Related to Innate Immune Responses in Ex Vivo Spinal Cord and Cerebellar Slice Cultures Infected with West Nile Virus. Brain Sci. 2018, 9, 1. [Google Scholar] [CrossRef]
- Salajegheh, M.; Kong, S.W.; Pinkus, J.L.; Walsh, R.J.; Liao, A.; Nazareno, R.; Amato, A.A.; Krastins, B.; Morehouse, C.; Higgs, B.W.; et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann. Neurol. 2010, 67, 53–63. [Google Scholar] [CrossRef]
- Marukian, S.; Andrus, L.; Sheahan, T.P.; Jones, C.T.; Charles, E.D.; Ploss, A.; Rice, C.M.; Dustin, L.B. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 2011, 54, 1913–1923. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Ysebrant de Lendonck, L.; Martinet, V.; Goriely, S. Interferon regulatory factor 3 in adaptive immune responses. Cell. Mol. Life Sci. 2014, 71, 3873–3883. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Negishi, H.; Taniguchi, T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, B.; Oh, G.T.; Kim, Y.J. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat. Immunol. 2013, 14, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Park, C.H.; Jeong, Y.H.; Kim, Y.J.; Ha, S.J. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8(+) T-cell exhaustion. PLoS Pathog. 2013, 9, e1003478. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Royer, W.E., Jr. Structural insights into interferon regulatory factor activation. Cell. Signal. 2010, 22, 883–887. [Google Scholar] [CrossRef]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ulrich, R.; Baumgärtner, W.; Gerhauser, I. Interferon-stimulated genes-essential antiviral effectors implicated in resistance to Theiler’s virus-induced demyelinating disease. J. Neuroinflamm. 2015, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Pine, R. Constitutive expression of an ISGF2/IRF1 transgene leads to interferon-independent activation of interferon-inducible genes and resistance to virus infection. J. Virol. 1992, 66, 4470–4478. [Google Scholar]
- Li, X.Q.; Li, X.N.; Liang, J.J.; Cai, X.B.; Tao, Q.; Li, Y.X.; Qin, Q.; Xu, S.P.; Luo, T.R. IRF1 up-regulates isg15 gene expression in dsRNA stimulation or CSFV infection by targeting nucleotides-487 to -325 in the 5’ flanking region. Mol. Immunol. 2018, 94, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.; Nemec, P.S.; Kapatos, A.; Miller, K.R.; Holmes, J.C.; Suter, S.E.; Buntzman, A.S.; Soderblom, E.J.; Collins, E.J.; Hess, P.R. The canine MHC class Ia allele DLA-88*508:01 presents diverse self- and canine distemper virus-origin peptides of varying length that have a conserved binding motif. Vet. Immunol. Immunopathol. 2018, 197, 76–86. [Google Scholar] [CrossRef]
- Graumann, M.B.; DeRose, S.A.; Ostrander, E.A.; Storb, R. Polymorphism analysis of four canine MHC class I genes. Tissue Antigens 1998, 51, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Mordstein, M.; Kochs, G.; Garcia-Sastre, A.; Tenoever, B.R. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 2010, 285, 42013–42022. [Google Scholar] [CrossRef]
- Schulz, K.S.; Mossman, K.L. Viral Evasion Strategies in Type I IFN Signaling—A Summary of Recent Developments. Front. Immunol. 2016, 7, 498. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Guerra, S.; Sanchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.F.; Mansur, D.S. Beyond ISGlylation: Functions of Free Intracellular and Extracellular ISG15. J. Interferon Cytokine Res. 2017, 37, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Bogunovic, D. ISG15: In Sickness and in Health. Trends Immunol. 2017, 38, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.X.; Yang, K.; Liu, X.; Liu, X.Y.; Wei, B.; Shan, Y.F.; Zhu, L.H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Speer, S.D.; Li, Z.; Buta, S.; Payelle-Brogard, B.; Qian, L.; Vigant, F.; Rubino, E.; Gardner, T.J.; Wedeking, T.; Hermann, M.; et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 2016, 7, 11496. [Google Scholar] [CrossRef]
- Bogunovic, D.; Byun, M.; Durfee, L.A.; Abhyankar, A.; Sanal, O.; Mansouri, D.; Salem, S.; Radovanovic, I.; Grant, A.V.; Adimi, P.; et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 2012, 337, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Recht, M.; Borden, E.C.; Knight, E., Jr. A human 15-kDa IFN-induced protein induces the secretion of IFN-gamma. J. Immunol. 1991, 147, 2617–2623. [Google Scholar] [PubMed]
- Zhang, Y.; Zhu, M.; Li, G.; Liu, J.; Zhai, X.; Wang, R.; Zhang, J.; Xing, G.; Gu, J.; Yan, L.; et al. Identification and function analysis of canine stimulator of interferon gene (STING). Microb. Pathog. 2017, 113, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Beineke, A.; Markus, S.; Borlak, J.; Thum, T.; Baumgärtner, W. Increase of pro-inflammatory cytokine expression in non-demyelinating early cerebral lesions in nervous canine distemper. Viral Immunol. 2008, 21, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Frisk, A.L.; Baumgärtner, W.; Gröne, A. Dominating interleukin-10 mRNA expression induction in cerebrospinal fluid cells of dogs with natural canine distemper virus induced demyelinating and non-demyelinating CNS lesions. J. Neuroimmunol. 1999, 97, 102–109. [Google Scholar] [CrossRef]
- Markus, S.; Failing, K.; Baumgärtner, W. Increased expression of pro-inflammatory cytokines and lack of up-regulation of anti-inflammatory cytokines in early distemper CNS lesions. J. Neuroimmunol. 2002, 125, 30–41. [Google Scholar] [CrossRef]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef]
- Marchal, J.A.; Lopez, G.J.; Peran, M.; Comino, A.; Delgado, J.R.; Garcia-Garcia, J.A.; Conde, V.; Aranda, F.M.; Rivas, C.; Esteban, M.; et al. The impact of PKR activation: From neurodegeneration to cancer. FASEB J. 2014, 28, 1965–1974. [Google Scholar] [CrossRef]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Elia, A. The double-stranded RNA-dependent protein kinase PKR: Structure and function. J. Interferon Cytokine Res. 1997, 17, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Yang, Y.L.; Weissmann, C.; Williams, B.R. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3279–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Danley, M.M.; LeVine, S.M. Immunohistochemical localization of phosphorylated protein kinase R and phosphorylated eukaryotic initiation factor-2 alpha in the central nervous system of SJL mice with experimental allergic encephalomyelitis. J. Neurosci. Res. 2004, 76, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: The 2’-5’oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Galabru, J.; Robert, N.; Buffet-Janvresse, C.; Riviere, Y.; Hovanessian, A.G. Continuous production of interferon in normal mice: Effect of anti-interferon globulin, sex, age, strain and environment on the levels of 2-5A synthetase and p67K kinase. J. Gen. Virol. 1985, 66 Pt 4, 711–718. [Google Scholar] [CrossRef]
- Flenniken, A.M.; Galabru, J.; Rutherford, M.N.; Hovanessian, A.G.; Williams, B.R. Expression of interferon-induced genes in different tissues of mice. J. Virol. 1988, 62, 3077–3083. [Google Scholar]
- Silverman, R.H. Fascination with 2-5A-dependent RNase: A unique enzyme that functions in interferon action. J. Interferon Res. 1994, 14, 101–104. [Google Scholar] [CrossRef]
- Iordanov, M.S.; Paranjape, J.M.; Zhou, A.; Wong, J.; Williams, B.R.; Meurs, E.F.; Silverman, R.H.; Magun, B.E. Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: Involvement of RNase L, protein kinase R, and alternative pathways. Mol. Cell. Biol. 2000, 20, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, J.; Wood, K.A.; Youle, R.J. The 2-5A system in viral infection and apoptosis. Biomed. Pharmacother. 1998, 52, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, H.; Scherer, C.A.; McVean, M.; Iadonato, S.P.; Vends, S.; Thavachelvam, K.; Steffensen, T.B.; Horan, K.A.; Kuri, T.; Weber, F.; et al. Extracellular 2’-5’ oligoadenylate synthetase stimulates RNase L-independent antiviral activity: A novel mechanism of virus-induced innate immunity. J. Virol. 2010, 84, 11898–11904. [Google Scholar] [CrossRef] [PubMed]
- Sorgeloos, F.; Jha, B.K.; Silverman, R.H.; Michiels, T. Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS Pathog. 2013, 9, e1003474. [Google Scholar] [CrossRef]
- Zhao, L.; Birdwell, L.D.; Wu, A.; Elliott, R.; Rose, K.M.; Phillips, J.M.; Li, Y.; Grinspan, J.; Silverman, R.H.; Weiss, S.R. Cell-type-specific activation of the oligoadenylate synthetase-RNase L pathway by a murine coronavirus. J. Virol. 2013, 87, 8408–8418. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Romeo, G.; Nissim, A.; Marziali, G.; Albertini, R.; Affabris, E.; Battistini, A.; Fiorucci, G.; Orsatti, R.; Rossi, G.B.; et al. A full-length murine 2-5A synthetase cDNA transfected in NIH-3T3 cells impairs EMCV but not VSV replication. Virology 1990, 179, 228–233. [Google Scholar] [CrossRef]
- Chebath, J.; Benech, P.; Revel, M.; Vigneron, M. Constitutive expression of (2’-5’) oligo A synthetase confers resistance to picornavirus infection. Nature 1987, 330, 587–588. [Google Scholar] [CrossRef] [PubMed]
- Rysiecki, G.; Gewert, D.R.; Williams, B.R. Constitutive expression of a 2’,5’-oligoadenylate synthetase cDNA results in increased antiviral activity and growth suppression. J. Interferon Res. 1989, 9, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Kochs, G. Interferon-induced Mx proteins in antiviral host defense. Biochimie 2007, 89, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Alldinger, S.; Baumgärtner, W.; Örvell, C. Restricted expression of viral surface proteins in canine distemper encephalitis. Acta Neuropathol. 1993, 85, 635–645. [Google Scholar] [CrossRef]
- Mutinelli, F.; Vandevelde, M.; Griot, C.; Richard, A. Astrocytic infection in canine distemper virus-induced demyelination. Acta Neuropathol. 1989, 77, 333–335. [Google Scholar] [CrossRef]
- Zurbriggen, A.; Schmid, I.; Graber, H.U.; Vandevelde, M. Oligodendroglial pathology in canine distemper. Acta Neuropathol. 1998, 95, 71–77. [Google Scholar] [CrossRef]
- Seehusen, F.; Baumgärtner, W. Axonal pathology and loss precede demyelination and accompany chronic lesions in a spontaneously occurring animal model of multiple sclerosis. Brain Pathol. 2010, 20, 551–559. [Google Scholar] [CrossRef]
- Vandevelde, M.; Zurbriggen, A. The neurobiology of canine distemper virus infection. Vet. Microbiol. 1995, 44, 271–280. [Google Scholar] [CrossRef]
- Gröters, S.; Alldinger, S.; Baumgärtner, W. Up-regulation of mRNA for matrix metalloproteinases-9 and -14 in advanced lesions of demyelinating canine distemper leukoencephalitis. Acta Neuropathol. 2005, 110, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Baumgärtner, W.; Failing, K.; Alldinger, S. Phase-dependent expression of matrix metalloproteinases and their inhibitors in demyelinating canine distemper encephalitis. Acta Neuropathol. 2003, 106, 486–494. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Gröne, A.; Alldinger, S.; Baumgärtner, W. Interleukin-1beta, -6, -12 and tumor necrosis factor-alpha expression in brains of dogs with canine distemper virus infection. J. Neuroimmunol. 2000, 110, 20–30. [Google Scholar] [CrossRef]
- Tipold, A.; Moore, P.; Zurbriggen, A.; Burgener, I.; Barben, G.; Vandevelde, M. Early T cell response in the central nervous system in canine distemper virus infection. Acta Neuropathol. 1999, 97, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wünschmann, A.; Alldinger, S.; Kremmer, E.; Baumgärtner, W. Identification of CD4+ and CD8+ T cell subsets and B cells in the brain of dogs with spontaneous acute, subacute-, and chronic-demyelinating distemper encephalitis. Vet. Immunol. Immunopathol. 1999, 67, 101–116. [Google Scholar] [CrossRef]
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 2018, 200, 1489–1495. [Google Scholar] [CrossRef]
- McDonough, A.; Lee, R.V.; Weinstein, J.R. Microglial Interferon Signaling and White Matter. Neurochem. Res. 2017, 42, 2625–2638. [Google Scholar] [CrossRef] [PubMed]
- Sanceau, J.; Boyd, D.D.; Seiki, M.; Bauvois, B. Interferons inhibit tumor necrosis factor-alpha-mediated matrix metalloproteinase-9 activation via interferon regulatory factor-1 binding competition with NF-kappa B. J. Biol. Chem. 2002, 277, 35766–35775. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, D.; Tamura, T.; Yanai, H.; Taniguchi, T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 2010, 59, 489–510. [Google Scholar] [CrossRef]
- Sweeney, S.E. Targeting interferon regulatory factors to inhibit activation of the type I IFN response: Implications for treatment of autoimmune disorders. Cell. Immunol. 2011, 271, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, S.E.; Kimbler, T.B.; Firestein, G.S. Synoviocyte innate immune responses: II. Pivotal role of IFN regulatory factor 3. J. Immunol. 2010, 184, 7162–7168. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Lee, D.H.; Oh, J.H.; Kim, M.K.; Kim, K.H.; Park, C.H.; Chung, J.H. Poly(I:C) induces expressions of MMP-1, -2, and -3 through various signaling pathways including IRF3 in human skin fibroblasts. J. Dermatol. Sci. 2015, 80, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Bienias, M.; Bruck, N.; Griep, C.; Wolf, C.; Kretschmer, S.; Kind, B.; Tungler, V.; Berner, R.; Lee-Kirsch, M.A. Therapeutic Approaches to Type I Interferonopathies. Curr. Rheumatol. Rep. 2018, 20, 32. [Google Scholar] [CrossRef]
- Lee-Kirsch, M.A. The Type I Interferonopathies. Annu. Rev. Med. 2017, 68, 297–315. [Google Scholar] [CrossRef]
- Rodero, M.P.; Crow, Y.J. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 2016, 213, 2527–2538. [Google Scholar] [CrossRef]
- Alldinger, S.; Wünschmann, A.; Baumgärtner, W.; Voss, C.; Kremmer, E. Up-regulation of major histocompatibility complex class II antigen expression in the central nervous system of dogs with spontaneous canine distemper virus encephalitis. Acta Neuropathol. 1996, 92, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.X.; Fish, E.N. The yin and yang of viruses and interferons. Trends Immunol 2012, 33, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.; Bihouee, A.; Teusan, R.; Dubois, E.; Savagner, F.; Steenman, M.; Houlgatte, R.; Ramstein, G. MADGene: Retrieval and processing of gene identifier lists for the analysis of heterogeneous microarray datasets. Bioinformatics 2011, 27, 725–726. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Acute | Subacute | Chronic |
|---|---|---|---|
| Pattern recognition receptors | |||
| Eif2ak2 (PKR) | 17.71 * | 15.62 * | 6.90 * |
| Ifih1 (MDA5) | 22.26 * | 33.76 * | 20.76 * |
| Tlr2 | 2.10 * | 2.84 * | 4.52 * |
| Tlr3 | 4.76 * | 5.87 * | 8.00 * |
| Tlr7 | 2.87 * | 2.83 * | 2.60 * |
| Interferon regulatory factors | |||
| Irf1 | 14.73 * | 30.13 * | 13.98 * |
| Irf3 | 1.17 | 1.23 | 1.32 |
| Irf5 | 1.06 | 1.03 | 1.71 |
| Irf7 | 45.42 * | 113.54 * | 24.65 * |
| Nfkb1 | 1.23 | 1.59 * | 1.39 |
| Type I interferons | |||
| Ifna1 | 1.00 | 1.00 | 1.00 |
| Ifnb1 | −1.00 | 1.01 | −1.00 |
| Type I interferon receptors | |||
| Ifnar1 | 1.33 | 1.53 * | −1.14 |
| Ifnar2 | 1.21 | 1.52 * | 1.25 |
| Signal transducers | |||
| Irf9 | 8.16 * | 11.44 * | 3.38 * |
| Jak1 | 1.12 | −1.27 | −1.22 |
| Socs1 | 1.00 | 1.00 | 1.00 |
| Stat1 | 12.03 * | 19.19 * | 7.12 * |
| Stat2 | 3.21 * | 4.57 * | 2.63 * |
| Tyk2 | 1.00 | 1.00 | 1.00 |
| Interferon-stimulated genes | |||
| IFI44 | 69.67 * | 78.05 * | 23.72 * |
| Ifi44l | 275.63 * | 419.93 * | 109.23 * |
| Ifit1 (Isg56) | 93.57 * | 179.05 * | 47.07 * |
| Ifit2 (Isg54) | 39.89 * | 77.21 * | 25.89 * |
| Isg15 | 590.52 * | 928.35 * | 245.71 * |
| Isg20 | 4.62 * | 8.05 * | 2.87 |
| Mx1 | 11.44 * | 15.98 * | 7.35 * |
| Mx2 | 47.68 * | 147.59 * | 38.43 * |
| Oas1 | 42.18 * | 71.15 * | 10.97 * |
| Oas2 | 80.24 * | 173.57 * | 34.76 * |
| Oasl | 44.47 * | 118.26 * | 33.62 * |
| Oasl2 | 4.42 * | 8.14 * | 2.61 * |
| Rnasel | 5.88 * | 9.07 * | 3.29 * |
| Major histocompatibility genes class I/II | |||
| DLA-12 (MHC I) | 6.72 * | 10.64 * | 4.94 * |
| DLA-64 (MHC I) | 24.52 * | 37.51 * | 18.07 * |
| DLA-79 (MHC I) | 10.49 * | 36.99 * | 9.17 * |
| DLA-88 (MHC I) | 17.87 * | 135.55 * | −4.80 |
| DLA-DQA1 (MHC II) | 12.19 * | 30.97 * | 17.87 * |
| DLA-DQB1 (MHC II) | 13.34 * | 22.98 * | 14.09 * |
| DLA-DRA (MHC II) | 5.27 * | 8.34 * | 6.78 * |
| DLA-DRB1 (MHC II) | 6.65 * | 9.77 * | 7.64 * |
| IRF3 | IRF7 | STAT1 | STAT2 | ISG15 | MX | OAS | PKR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDV | + | - | + | - | + | - | + | - | + | - | + | - | + | - | + | - |
| Neurons | + | + | ++ | ++ | + | (+) | ++ | ++ | ++ | + | +++ | + | ++ | ++ | ++ | ++ |
| Purkinje cells | + | (+) | (+) | (+) | + | (+) | ++ | ++ | ++ | + | +++ | - | ++ | ++ | +++ | +++ |
| Granular cells | + | + | ++ | ++ | - | - | - | - | + | - | +++ | - | (+) | (+) | - | - |
| Glial cells | ++ | ++ | ++ | ++ | +++ | + | +++ | ++ | + | - | +++ | - | + | - | ++ | - |
| Endothelial cells | +++ | +++ | (+) | (+) | + | (+) | - | - | ++ | (+) | ++ | (+) | + | + | (+) | - |
| Inflammatory cells | +++ | n.a. | - | n.a. | + | n.a. | - | n.a. | - | n.a. | ++ | n.a. | ++ | n.a. | +++ | n.a. |
| CDV | IRF3 | IRF7 | STAT1 | STAT2 | ISG15 | MX | OAS | PKR | |
|---|---|---|---|---|---|---|---|---|---|
| CDV | . | 0.205* | 0.026 | 0.541 * | 0.259 * | 0.490 * | 0.594 * | 0.289 * | 0.454 * |
| IRF3 | 0.205 * | . | −0.034 | 0.162 * | 0.034 | 0.052 | −0.063 | 0.204 * | 0.249 * |
| IRF7 | 0.026 | −0.034 | . | 0.334 * | 0.146 * | 0.404 * | 0.332 * | 0.268 * | 0.225 * |
| STAT1 | 0.541 * | 0.162* | 0.334 * | . | 0.357 * | 0.442 * | 0.586 * | 0.404 * | 0.436 * |
| STAT2 | 0.259 * | 0.034 | 0.146 * | 0.357 * | . | 0.225 * | 0.314 * | 0.174 * | 0.256 * |
| ISG15 | 0.490 * | 0.052 | 0.404 * | 0.442 * | 0.225 * | . | 0.640 * | 0.454 * | 0.471 * |
| MX | 0.594 * | −0.063 | 0.332 * | 0.586 * | 0.314 * | 0.640 * | . | 0.423 * | 0.520 * |
| OAS | 0.289 * | 0.204 * | 0.268 * | 0.404 * | 0.174 * | 0.454 * | 0.423 * | . | 0.331 * |
| PKR | 0.454 * | 0.249 * | 0.225 * | 0.436 * | 0.256 * | 0.471 * | 0.520 * | 0.331 * | . |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klotz, D.; Gerhauser, I. Interferon-Stimulated Genes—Mediators of the Innate Immune Response during Canine Distemper Virus Infection. Int. J. Mol. Sci. 2019, 20, 1620. https://doi.org/10.3390/ijms20071620
Klotz D, Gerhauser I. Interferon-Stimulated Genes—Mediators of the Innate Immune Response during Canine Distemper Virus Infection. International Journal of Molecular Sciences. 2019; 20(7):1620. https://doi.org/10.3390/ijms20071620
Chicago/Turabian StyleKlotz, Daniela, and Ingo Gerhauser. 2019. "Interferon-Stimulated Genes—Mediators of the Innate Immune Response during Canine Distemper Virus Infection" International Journal of Molecular Sciences 20, no. 7: 1620. https://doi.org/10.3390/ijms20071620
APA StyleKlotz, D., & Gerhauser, I. (2019). Interferon-Stimulated Genes—Mediators of the Innate Immune Response during Canine Distemper Virus Infection. International Journal of Molecular Sciences, 20(7), 1620. https://doi.org/10.3390/ijms20071620