Molecular Mechanisms Controlled by mTOR in Male Reproductive System




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
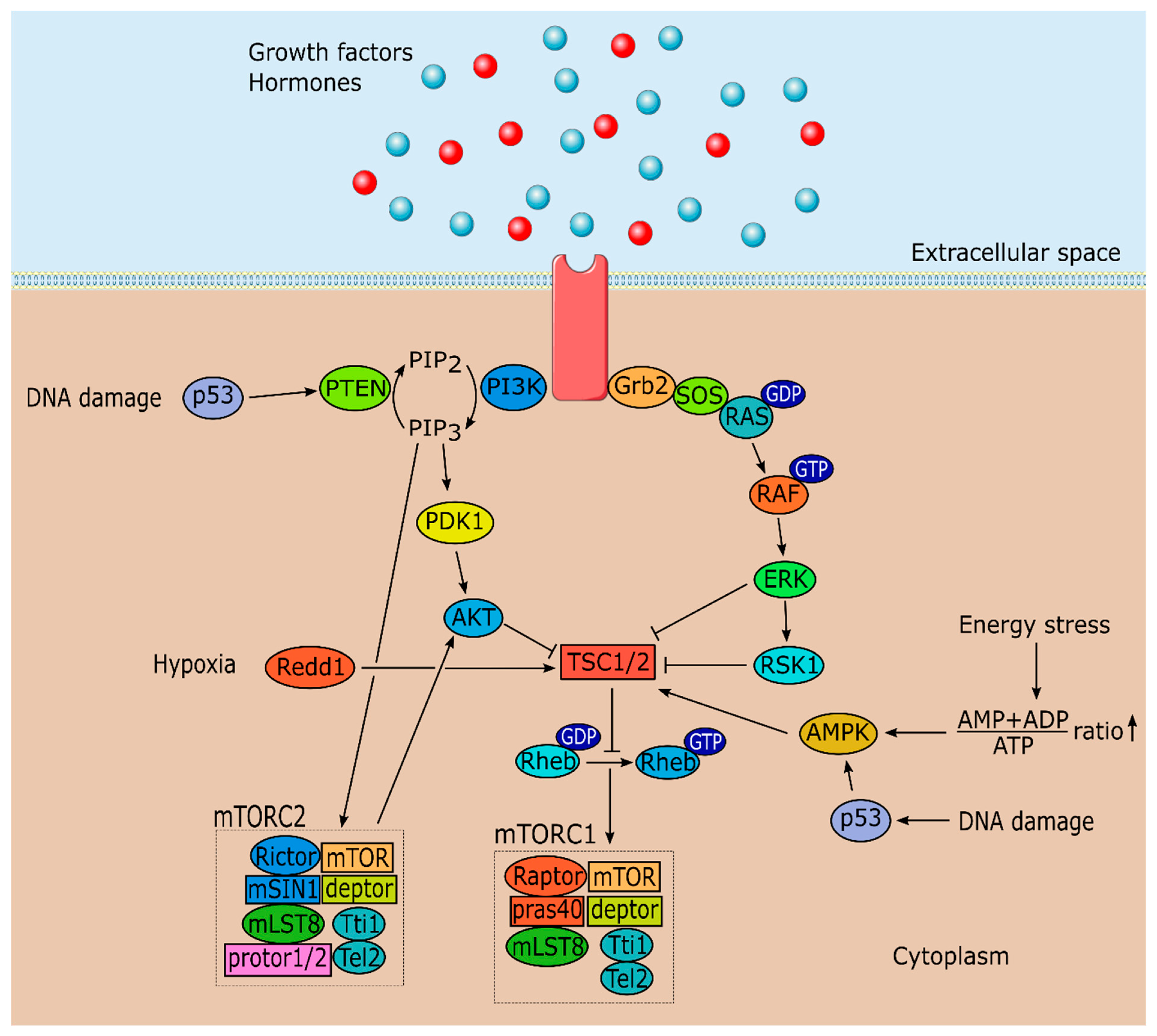
2. mTOR Signaling and Cell Physiology: Brief Overview
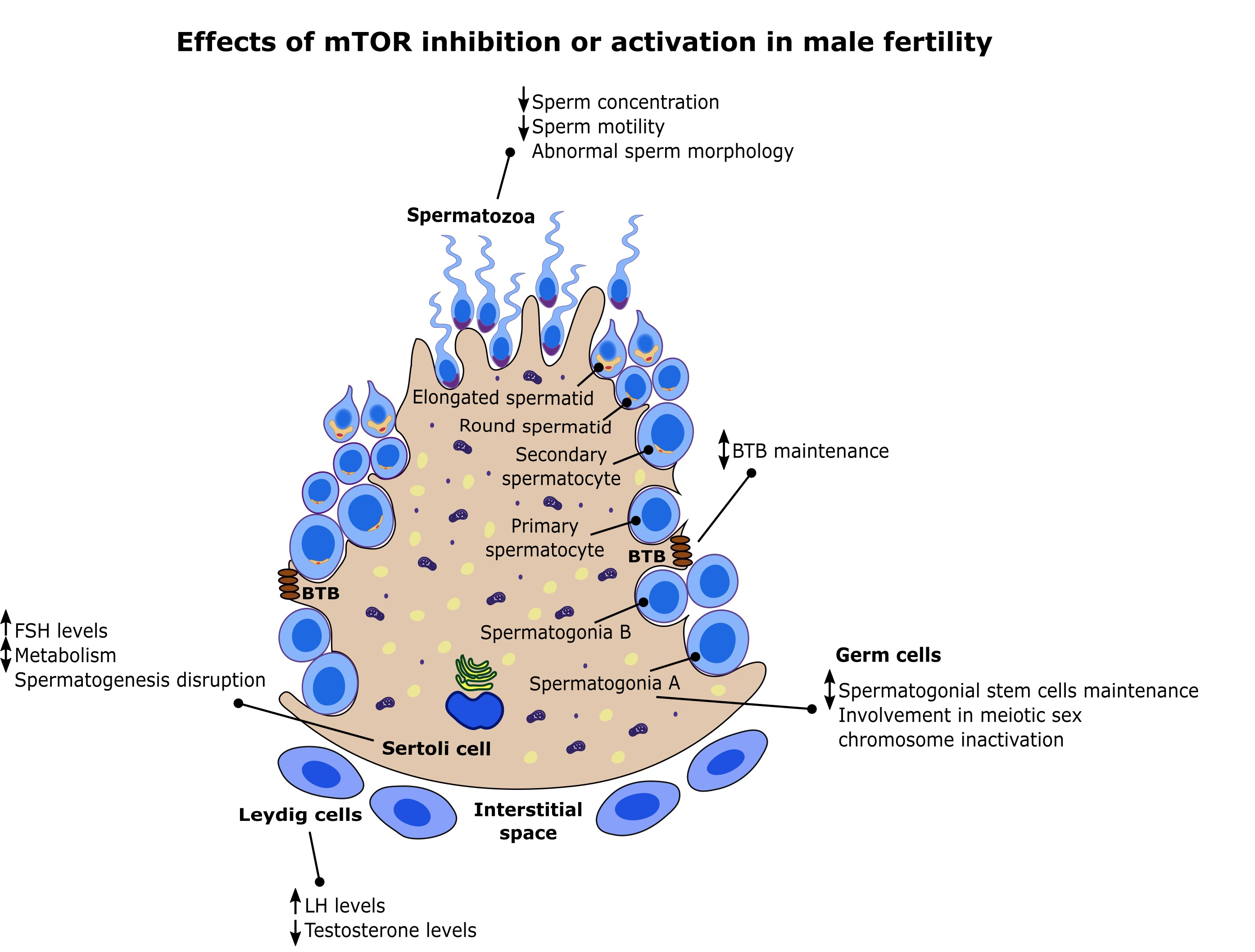
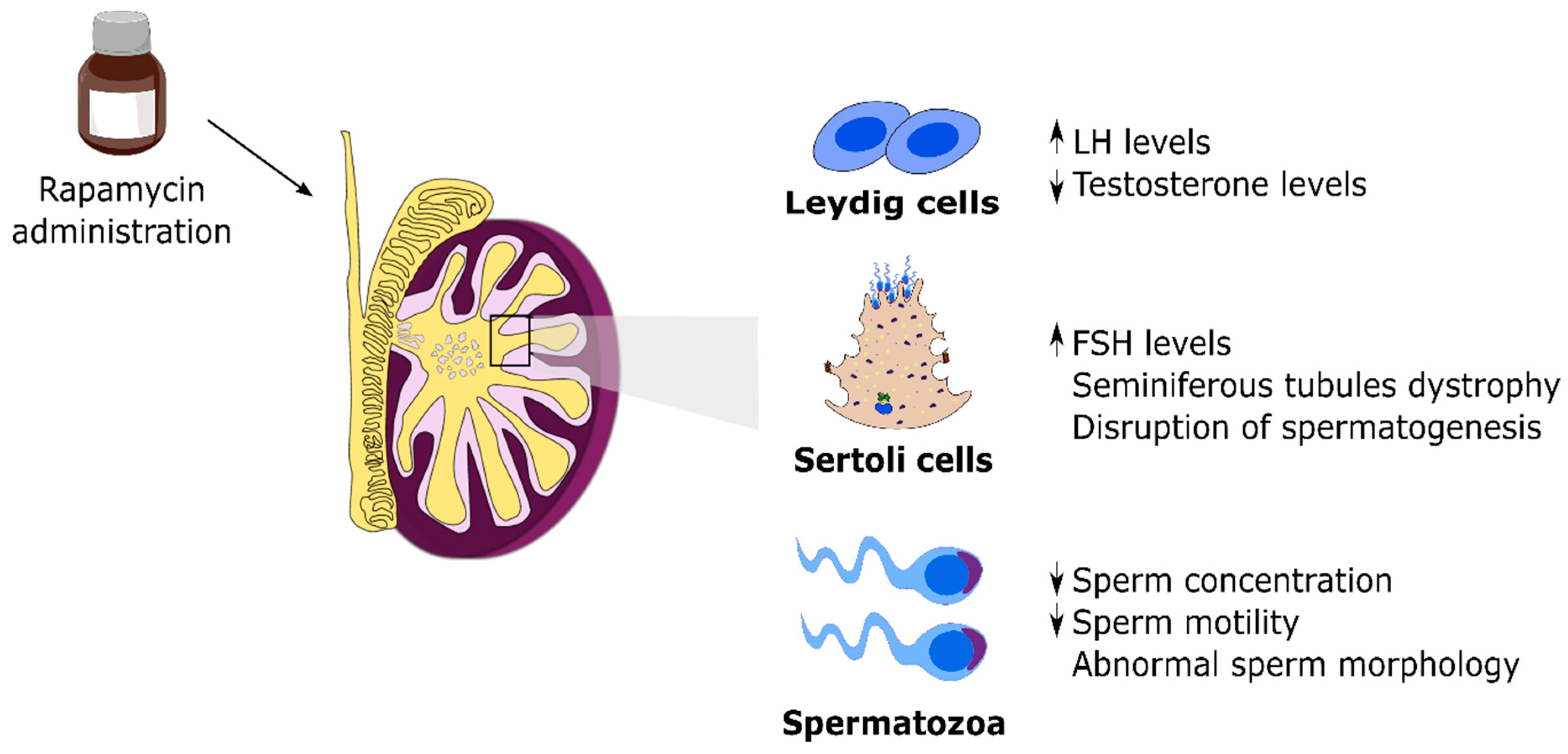
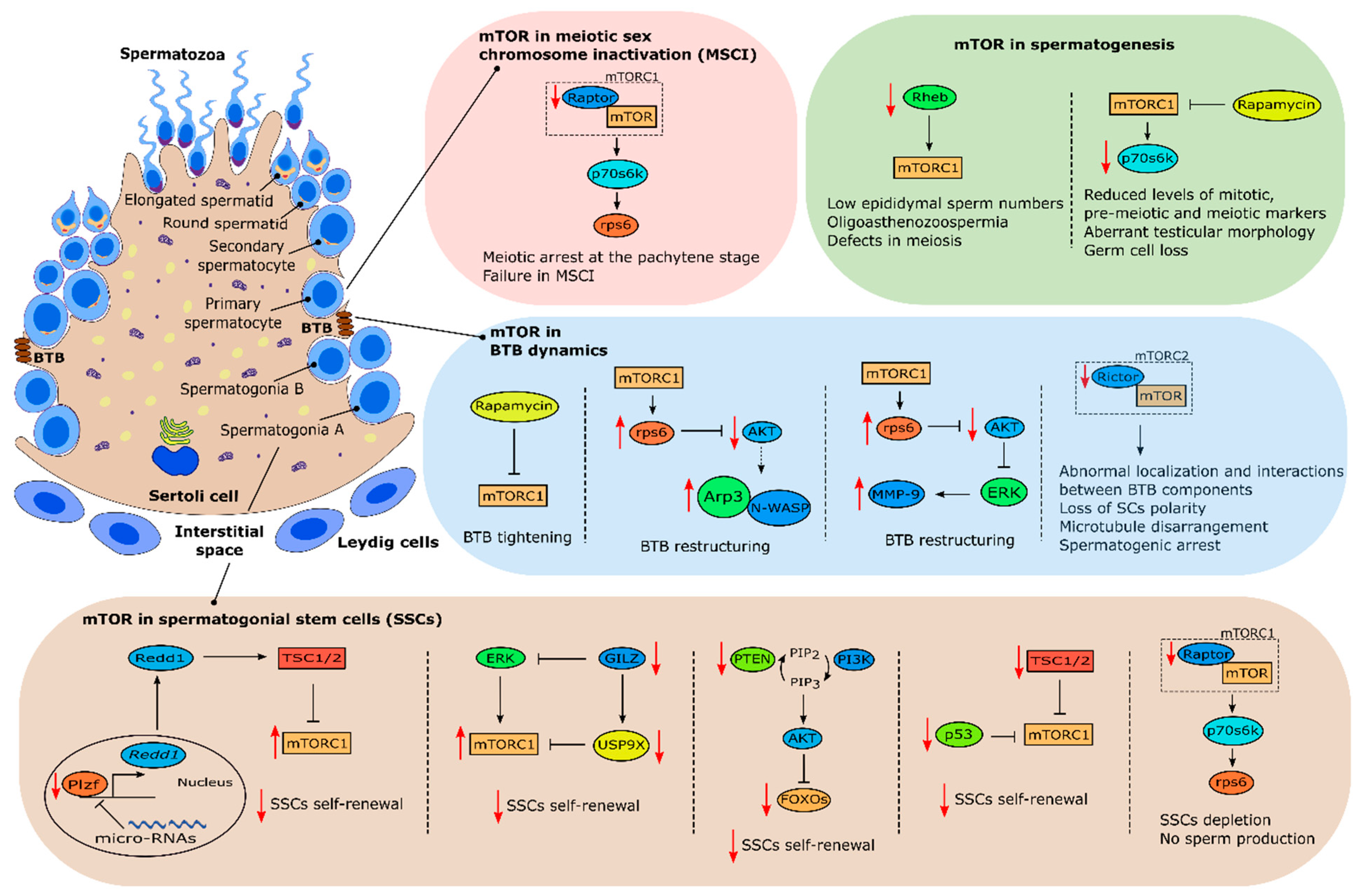
3. mTOR and Male Fertility: Evidence from Testis Signaling
4. mTOR Pathway in Sertoli Cells and Male Fertility
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mTOR | Mammalian target of rapamycin |
| SSCs | Spermatogonial stem cells |
| SCs | Sertoli cells |
| mTORC1 | Mammalian target of rapamycin 1 |
| mTORC2 | Mammalian target of rapamycin 2 |
| BTB | Blood-testis barrier |
| raptor | Regulatory associated protein of mTOR |
| pras40 | Proline-rich Akt substrate 40 kDa |
| deptor | DEP (Dishevelled, Egl-10 and Pleckstrin) domain-containing mTOR-interacting protein |
| mLST8 | Mammalian lethal with sec-13 protein 8 |
| rictor | Rapamycin insensitive companion of mTOR |
| mSIN1 | Mammalian stress-activated protein kinase interacting protein |
| protor1/2 | Protein observed with rictor 1 and 2 |
| FKBP12 | FK506-binding protein 12 |
| TSC1/2 | Tuberous sclerosis complex |
| Rheb | Ras homolog enriched in brain GTPase |
| Akt | Protein kinase B |
| RSK1 | p90 ribosomal S6 kinase 1 |
| ERK | Extracellular signal regulated kinase |
| PTEN | Phosphatase and tensin homolog |
| Redd1 | Protein regulated in development and DNA damage response 1 |
| PKC | Protein kinase C |
| Plzf | Promyelocytic leukaemia zinc finger |
| GNDF | Glial cell-derived neurotrophic factor |
| FOXOs | Forkhead box proteins |
| GILZ | Glucocorticoid-induced leucine zipper |
| USP9X | Spermatogonial deubiquitinase probable ubiquitin carboxyl-terminal hydrolase FAF-X |
| STRA8 | Retinoic acid stimulated gene 8 |
| p-p70s6k | Phosphorylated p70S6 kinase |
| p-4E-BP1 | Phosphorylated eukaryotic initiation factor 4E binding protein 1 |
| PCNA | Proliferating cell nuclear antigen |
| Dmc1 | DNA meiotic recombinase 1 |
| Rps6 | Ribosomal protein S6 |
| MSCI | Meiotic sex chromosome inactivation |
| ZO-1 | Zonula occludens-1 |
| N-WASP | Neuronal Wiskott-Aldrich syndrome protein |
| MMP-9 | Matrix metallopeptidase 9 |
| ARHGEF4 | Rho guanine nucleotide exchange factor 4 |
| GLP-1 | Glucagon-like peptide-1 |
| AMPK | AMP-activated protein kinase |
References
- Cannon, W.B. Physiological Regulation of Normal States: Some Tentative Postulates Concerning Biological Homeostatics; éditions Médicales: Paris, France, 1926. [Google Scholar]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Mtor interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (tor), mediates tor action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mtor, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian tor complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. Raft1: A mammalian protein that binds to fkbp12 in a rapamycin-dependent fashion and is homologous to yeast tors. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Jesus, T.T.; Oliveira, P.F.; Silva, J.; Barros, A.; Ferreira, R.; Sousa, M.; Cheng, C.Y.; Silva, B.M.; Alves, M.G. Mammalian target of rapamycin controls glucose consumption and redox balance in human sertoli cells. Fertil. Steril. 2016, 105, 825–833. [Google Scholar] [CrossRef]
- Mok, K.W.; Chen, H.; Lee, W.M.; Cheng, C.Y. Rps6 regulates blood-testis barrier dynamics through arp3-mediated actin microfilament organization in rat sertoli cells. An in vitro study. Endocrinology 2015, 156, 1900–1913. [Google Scholar] [CrossRef]
- Mok, K.W.; Mruk, D.D.; Cheng, C.Y. Rps6 regulates blood-testis barrier dynamics through akt-mediated effects on mmp-9. J. Cell Sci. 2014, 127, 4870–4882. [Google Scholar] [CrossRef] [PubMed]
- Mok, K.W.; Mruk, D.D.; Lee, W.M.; Cheng, C.Y. Rictor/mtorc2 regulates blood-testis barrier dynamics via its effects on gap junction communications and actin filament network. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1137–1152. [Google Scholar] [CrossRef]
- Rato, L.; Alves, M.G.; Socorro, S.; Duarte, A.I.; Cavaco, J.E.; Oliveira, P.F. Metabolic regulation is important for spermatogenesis. Nat. Rev. Urol. 2012, 9, 330–338. [Google Scholar] [CrossRef]
- Hess, R.A.; Renato de Franca, L. Spermatogenesis and cycle of the seminiferous epithelium. Adv. Exp. Med. Biol. 2008, 636, 1–15. [Google Scholar]
- Rato, L.; Meneses, M.J.; Silva, B.M.; Sousa, M.; Alves, M.G.; Oliveira, P.F. New insights on hormones and factors that modulate sertoli cell metabolism. Histol. Histopathol. 2016, 31, 499–513. [Google Scholar]
- Boussouar, F.; Benahmed, M. Lactate and energy metabolism in male germ cells. Trends Endocrinol. Metab. 2004, 15, 345–350. [Google Scholar] [CrossRef]
- Mruk, D.D.; Cheng, C.Y. The mammalian blood-testis barrier: Its biology and regulation. Endocr. Rev. 2015, 36, 564–591. [Google Scholar] [CrossRef]
- Dong, H.; Chen, Z.; Wang, C.; Xiong, Z.; Zhao, W.; Jia, C.; Lin, J.; Lin, Y.; Yuan, W.; Zhao, A.Z.; et al. Rictor regulates spermatogenesis by controlling sertoli cell cytoskeletal organization and cell polarity in the mouse testis. Endocrinology 2015, 156, 4244–4256. [Google Scholar] [CrossRef]
- Mok, K.W.; Mruk, D.D.; Silvestrini, B.; Cheng, C.Y. Rps6 regulates blood-testis barrier dynamics by affecting f-actin organization and protein recruitment. Endocrinology 2012, 153, 5036–5048. [Google Scholar] [CrossRef]
- Libby, E.; Ratcliff, W.C. Evolution. Ratcheting the evolution of multicellularity. Science 2014, 346, 426–427. [Google Scholar] [CrossRef]
- Dussutour, A.; Latty, T.; Beekman, M.; Simpson, S.J. Amoeboid organism solves complex nutritional challenges. Proc. Natl. Acad. Sci. USA 2010, 107, 4607–4611. [Google Scholar] [CrossRef] [Green Version]
- Szathmáry, E.; Smith, J.M. The major evolutionary transitions. Nature 1995, 374, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. Pras40 is an insulin-regulated inhibitor of the mtorc1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jeno, P.; Arrieumerlou, C.; Hall, M.N. Pras40 and prr5-like protein are new mtor interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mtor inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mtor complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Boil. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. Msin1 is necessary for akt/pkb phosphorylation, and its isoforms define three distinct mtorc2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of protor as a novel rictor-binding component of mtor complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (ay-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Martel, R.R.; Klicius, J.; Galet, S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can. J. Physiol. Pharmacol. 1977, 55, 48–51. [Google Scholar] [CrossRef]
- Eng, C.P.; Sehgal, S.N.; Vezina, C. Activity of rapamycin (ay-22,989) against transplanted tumors. J. Antibiot. 1984, 37, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, tuberin and hamartin, control mtor signaling by acting as a gtpase-activating protein complex toward rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mtor kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The dawn of the age of amino acid sensors for the mtorc1 pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef]
- Bröer, S.; Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the tsc complex integrates insulin and nutrient regulation of mtorc1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Randle, S.C. Tuberous sclerosis complex: A review. Pediatr. Ann. 2017, 46, e166–e171. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of tsc2 by erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. Tsc2 is phosphorylated and inhibited by akt and suppresses mtor signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal s6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. Ampk phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of ampk beta1, tsc2, and pten expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the igf-1-akt-mtor pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mtor pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of pten transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mtor function in response to hypoxia by redd1 and the tsc1/tsc2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates tsc1/2-mtor signaling and tumor suppression through redd1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of sin1 as an essential torc2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, J.M.; Alessi, D.R. Mtor complex 2 (mtorc2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (sgk1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of akt/pkb by the rictor-mtor complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. Ptdins(3,4,5)p3-dependent activation of the mtorc2 kinase complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between akt and mtorc2 via sin1 phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The tsc1-tsc2 complex is required for proper activation of mtor complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mtorc2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Zhou, X.; Qian, J.; Zhu, W.; Shu, Y.; Liu, P. Clinical efficacy of mtor inhibitors in solid tumors: A systematic review. Future Oncol. 2015, 11, 1687–1699. [Google Scholar] [CrossRef]
- Fouque, A.; Jean, M.; Weghe, P.; Legembre, P. Review of pi3k/mtor inhibitors entering clinical trials to treat triple negative breast cancers. Recent Patents Anti-Cancer Drug Discov. 2016, 11, 283–296. [Google Scholar] [CrossRef]
- Ortolani, S.; Ciccarese, C.; Cingarlini, S.; Tortora, G.; Massari, F. Suppression of mtor pathway in solid tumors: Lessons learned from clinical experience in renal cell carcinoma and neuroendocrine tumors and new perspectives. Future Oncol. 2015, 11, 1809–1828. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Constanti, A.; Russo, E.; De Sarro, G. Mtor pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol. Res. 2016, 107, 333–343. [Google Scholar] [CrossRef]
- De Fijter, J.W. Cancer and mtor inhibitors in transplant recipients. Transplantation 2017, 101, 45–55. [Google Scholar] [CrossRef]
- Ghidini, M.; Petrelli, F.; Ghidini, A.; Tomasello, G.; Hahne, J.C.; Passalacqua, R.; Barni, S. Clinical development of mtor inhibitors for renal cancer. Expert Opin. Investig. Drugs 2017, 26, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Verges, B.; Cariou, B. Mtor inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef]
- Yoo, Y.J.; Kim, H.; Park, S.R.; Yoon, Y.J. An overview of rapamycin: From discovery to future perspectives. J. Ind. Microbiol. Biotechnol. 2017, 44, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, E.; Zairi, A.; Nohra, J.; Kamar, N.; Plante, P.; Rostaing, L. Gonadal impact of target of rapamycin inhibitors (sirolimus and everolimus) in male patients: An overview. Transpl. Int. Off. J. Eur. Soc. Organ Transplant. 2007, 20, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bererhi, L.; Flamant, M.; Martinez, F.; Karras, A.; Thervet, E.; Legendre, C. Rapamycin-induced oligospermia. Transplantation 2003, 76, 885–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, I.; Groetzner, J.; Adamidis, I.; Landwehr, P.; Mueller, M.; Vogeser, M.; Gerstorfer, M.; Uberfuhr, P.; Meiser, B.; Reichart, B. Sirolimus impairs gonadal function in heart transplant recipients. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2004, 4, 1084–1088. [Google Scholar] [CrossRef]
- Zuber, J.; Anglicheau, D.; Elie, C.; Bererhi, L.; Timsit, M.O.; Mamzer-Bruneel, M.F.; Ciroldi, M.; Martinez, F.; Snanoudj, R.; Hiesse, C.; et al. Sirolimus may reduce fertility in male renal transplant recipients. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2008, 8, 1471–1479. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.; Lin, Y.; Lin, H.; Li, M.; Nie, P.; Chen, L.; Qiu, J.; Lu, Y.; Chen, L.; et al. Long-term impact of immunosuppressants at therapeutic doses on male reproductive system in unilateral nephrectomized rats: A comparative study. BioMed Res. Int. 2013, 2013, 690382. [Google Scholar] [CrossRef]
- Rovira, J.; Diekmann, F.; Ramirez-Bajo, M.J.; Banon-Maneus, E.; Moya-Rull, D.; Campistol, J.M. Sirolimus-associated testicular toxicity: Detrimental but reversible. Transplantation 2012, 93, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, J.; Krause, W. Azoospermia in a renal transplant recipient during sirolimus (rapamycin) treatment. Andrologia 2007, 39, 198–199. [Google Scholar] [CrossRef]
- Hobbs, R.M.; Seandel, M.; Falciatori, I.; Rafii, S.; Pandolfi, P.P. Plzf regulates germline progenitor self-renewal by opposing mtorc1. Cell 2010, 142, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Costoya, J.A.; Hobbs, R.M.; Barna, M.; Cattoretti, G.; Manova, K.; Sukhwani, M.; Orwig, K.E.; Wolgemuth, D.J.; Pandolfi, P.P. Essential role of plzf in maintenance of spermatogonial stem cells. Nat. Genet. 2004, 36, 653–659. [Google Scholar] [CrossRef]
- Buaas, F.W.; Kirsh, A.L.; Sharma, M.; McLean, D.J.; Morris, J.L.; Griswold, M.D.; de Rooij, D.G.; Braun, R.E. Plzf is required in adult male germ cells for stem cell self-renewal. Nat. Genet. 2004, 36, 647–652. [Google Scholar] [CrossRef] [Green Version]
- Daguia Zambe, J.C.; Zhai, Y.; Zhou, Z.; Du, X.; Wei, Y.; Ma, F.; Hua, J. Mir-19b-3p induces cell proliferation and reduces heterochromatin-mediated senescence through plzf in goat male germline stem cells. J. Cell. Physiol. 2018, 233, 4652–4665. [Google Scholar] [CrossRef]
- Tarnawa, E.D.; Baker, M.D.; Aloisio, G.M.; Carr, B.R.; Castrillon, D.H. Gonadal expression of foxo1, but not foxo3, is conserved in diverse mammalian species. Biol. Reprod. 2013, 88, 103. [Google Scholar] [CrossRef] [PubMed]
- Goertz, M.J.; Wu, Z.; Gallardo, T.D.; Hamra, F.K.; Castrillon, D.H. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J. Clin. Investig. 2011, 121, 3456–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the igf-1/mtor pathways by the p53 protein. Trends Cell Boil. 2010, 20, 427–434. [Google Scholar] [CrossRef]
- Solozobova, V.; Blattner, C. P53 in stem cells. World J. Biol. Chem. 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Xiong, M.; Ferder, I.C.; Ohguchi, Y.; Wang, N. Quantitative analysis of male germline stem cell differentiation reveals a role for the p53-mtorc1 pathway in spermatogonial maintenance. Cell Cycle 2015, 14, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, R.M.; La, H.M.; Makela, J.A.; Kobayashi, T.; Noda, T.; Pandolfi, P.P. Distinct germline progenitor subsets defined through tsc2-mtorc1 signaling. EMBO Rep. 2015, 16, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Xiong, Z.; Dai, H.; Zou, Z.; Jia, C.; Bai, X.; Chen, Z. Mtorc1 activation promotes spermatogonial differentiation and causes subfertility in mice. Biol. Reprod. 2016, 95, 97. [Google Scholar] [CrossRef] [PubMed]
- Bruscoli, S.; Velardi, E.; Di Sante, M.; Bereshchenko, O.; Venanzi, A.; Coppo, M.; Berno, V.; Mameli, M.G.; Colella, R.; Cavaliere, A.; et al. Long glucocorticoid-induced leucine zipper (l-gilz) protein interacts with ras protein pathway and contributes to spermatogenesis control. J. Biol. Chem. 2012, 287, 1242–1251. [Google Scholar] [CrossRef]
- Ngo, D.; Cheng, Q.; O’Connor, A.E.; DeBoer, K.D.; Lo, C.Y.; Beaulieu, E.; De Seram, M.; Hobbs, R.M.; O’Bryan, M.K.; Morand, E.F. Glucocorticoid-induced leucine zipper (gilz) regulates testicular foxo1 activity and spermatogonial stem cell (ssc) function. PLoS ONE 2013, 8, e59149. [Google Scholar] [CrossRef]
- La, H.M.; Chan, A.L.; Legrand, J.M.D.; Rossello, F.J.; Gangemi, C.G.; Papa, A.; Cheng, Q.; Morand, E.F.; Hobbs, R.M. Gilz-dependent modulation of mtorc1 regulates spermatogonial maintenance. Development 2018, 145, dev165324. [Google Scholar] [CrossRef]
- Kishi, K.; Uchida, A.; Takase, H.M.; Suzuki, H.; Kurohmaru, M.; Tsunekawa, N.; Kanai-Azuma, M.; Wood, S.A.; Kanai, Y. Spermatogonial deubiquitinase usp9x is essential for proper spermatogenesis in mice. Reproduction 2017, 154, 135–143. [Google Scholar] [CrossRef]
- Meng, X.; Lindahl, M.; Hyvonen, M.E.; Parvinen, M.; de Rooij, D.G.; Hess, M.W.; Raatikainen-Ahokas, A.; Sainio, K.; Rauvala, H.; Lakso, M.; et al. Regulation of cell fate decision of undifferentiated spermatogonia by gdnf. Science 2000, 287, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Avarbock, M.R.; Brinster, R.L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16489–16494. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Guo, Y.; Wang, M.; Zhou, T.; Xue, Y.; Du, G.; Wei, X.; Wang, J.; Qi, L.; Zhang, H.; et al. The glial cell-derived neurotrophic factor (gdnf)-responsive phosphoprotein landscape identifies raptor phosphorylation required for spermatogonial progenitor cell proliferation. Mol. Cell. Proteom. 2017, 16, 982–997. [Google Scholar] [CrossRef] [PubMed]
- Serra, N.D.; Velte, E.K.; Niedenberger, B.A.; Kirsanov, O.; Geyer, C.B. Cell-autonomous requirement for mammalian target of rapamycin (mtor) in spermatogonial proliferation and differentiation in the mousedagger. Biol. Reprod. 2017, 96, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Busada, J.T.; Niedenberger, B.A.; Velte, E.K.; Keiper, B.D.; Geyer, C.B. Mammalian target of rapamycin complex 1 (mtorc1) is required for mouse spermatogonial differentiation in vivo. Dev. Biol. 2015, 407, 90–102. [Google Scholar] [CrossRef]
- Serra, N.; Velte, E.K.; Niedenberger, B.A.; Kirsanov, O.; Geyer, C.B. The mtorc1 component rptor is required for maintenance of the foundational spermatogonial stem cell pool in mice. Biol. Reprod. 2018, 100, 429–439. [Google Scholar] [CrossRef]
- Baker, M.D.; Ezzati, M.; Aloisio, G.M.; Tarnawa, E.D.; Cuevas, I.; Nakada, Y.; Castrillon, D.H. The small gtpase rheb is required for spermatogenesis but not oogenesis. Reproduction 2014, 147, 615–625. [Google Scholar]
- Sahin, P.; Gungor-Ordueri, N.E.; Celik-Ozenci, C. Inhibition of mtor pathway decreases the expression of pre-meiotic and meiotic markers throughout postnatal development and in adult testes in mice. Andrologia 2018, 50, e12811. [Google Scholar] [CrossRef]
- Xu, H.; Shen, L.; Chen, X.; Ding, Y.; He, J.; Zhu, J.; Wang, Y.; Liu, X. Mtor/p70s6k promotes spermatogonia proliferation and spermatogenesis in sprague dawley rats. Reprod. Biomed. Online 2016, 32, 207–217. [Google Scholar] [CrossRef]
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Nie, R.; Li, Y.; Friel, P.; Mitchell, D.; Hess, R.A.; Small, C.; Griswold, M.D. Expression of stimulated by retinoic acid gene 8 (stra8) in spermatogenic cells induced by retinoic acid: An in vivo study in vitamin a-sufficient postnatal murine testes. Biol. Reprod. 2008, 79, 35–42. [Google Scholar]
- Anderson, E.L.; Baltus, A.E.; Roepers-Gajadien, H.L.; Hassold, T.J.; de Rooij, D.G.; van Pelt, A.M.; Page, D.C. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 14976–14980. [Google Scholar] [CrossRef] [Green Version]
- Sahin, P.; Sahin, Z.; Gungor-Ordueri, N.E.; Donmez, B.O.; Celik-Ozenci, C. Inhibition of mammalian target of rapamycin signaling pathway decreases retinoic acid stimulated gene 8 expression in adult mouse testis. Fertil. Steril. 2014, 102, 1482–1490. [Google Scholar] [CrossRef]
- Turner, J.M. Meiotic sex chromosome inactivation. Development 2007, 134, 1823–1831. [Google Scholar] [PubMed] [Green Version]
- Fernandez-Capetillo, O.; Mahadevaiah, S.K.; Celeste, A.; Romanienko, P.J.; Camerini-Otero, R.D.; Bonner, W.M.; Manova, K.; Burgoyne, P.; Nussenzweig, A. H2ax is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell 2003, 4, 497–508. [Google Scholar] [CrossRef]
- Turner, J.M.; Mahadevaiah, S.K.; Ellis, P.J.; Mitchell, M.J.; Burgoyne, P.S. Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev. Cell 2006, 10, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Zhu, Z.; Tian, S.; Zhu, R.; Bai, S.; Fu, K.; Davis, J.G.; Sun, Z.; Baur, J.A.; Zheng, K.; et al. Conditional ablation of raptor in the male germline causes infertility due to meiotic arrest and impaired inactivation of sex chromosomes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3934–3949. [Google Scholar] [CrossRef]
- Bai, S.; Cheng, L.; Zhang, Y.; Zhu, C.; Zhu, Z.; Zhu, R.; Cheng, C.Y.; Ye, L.; Zheng, K. A germline-specific role for the mtorc2 component rictor in maintaining spermatogonial differentiation and intercellular adhesion in mouse testis. Mol. Hum. Reprod. 2018, 24, 244–259. [Google Scholar] [CrossRef]
- Zhu, Z.; Yue, Q.; Xie, J.; Zhang, S.; He, W.; Bai, S.; Tian, S.; Zhang, Y.; Xiong, M.; Sun, Z.; et al. Rapamycin-mediated mtor inhibition impairs silencing of sex chromosomes and the pachytene pirna pathway in the mouse testis. Aging 2019, 11, 185–208. [Google Scholar]
- Weber, J.E.; Russell, L.D.; Wong, V.; Peterson, R.N. Three-dimensional reconstruction of a rat stage v sertoli cell: Ii. Morphometry of sertoli–sertoli and sertoli–germ-cell relationships. Am. J. Anat. 1983, 167, 163–179. [Google Scholar] [CrossRef]
- Kaur, G.; Thompson, L.A.; Dufour, J.M. Sertoli cells--immunological sentinels of spermatogenesis. Semin. Cell Dev. Boil. 2014, 30, 36–44. [Google Scholar] [CrossRef]
- O’Donnell, L. Mechanisms of spermiogenesis and spermiation and how they are disturbed. Spermatogenesis 2014, 4, e979623. [Google Scholar] [CrossRef]
- Li, N.; Mruk, D.D.; Cheng, C.Y. Actin binding proteins in blood-testis barrier function. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.; Mruk, D.D.; Lee, W.M.; Cheng, C.Y. Cross-talk between tight and anchoring junctions-lesson from the testis. Adv. Exp. Med. Biol. 2008, 636, 234–254. [Google Scholar] [PubMed]
- Xiao, X.; Mruk, D.D.; Wong, C.K.; Cheng, C.Y. Germ cell transport across the seminiferous epithelium during spermatogenesis. Physiology 2014, 29, 286–298. [Google Scholar] [PubMed]
- Russell, L. Movement of spermatocytes from the basal to the adluminal compartment of the rat testis. Am. J. Anat. 1977, 148, 313–328. [Google Scholar] [CrossRef]
- Li, S.Y.T.; Yan, M.; Chen, H.; Jesus, T.; Lee, W.M.; Xiao, X.; Cheng, C.Y. Mtorc1/rps6 regulates blood-testis barrier dynamics and spermatogenetic function in the testis in vivo. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E174–E190. [Google Scholar]
- Xiong, Z.; Wang, C.; Wang, Z.; Dai, H.; Song, Q.; Zou, Z.; Xiao, B.; Zhao, A.Z.; Bai, X.; Chen, Z. Raptor directs sertoli cell cytoskeletal organization and polarity in the mouse testis. Biol. Reprod. 2018, 99, 1289–1302. [Google Scholar] [PubMed]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mtorc1 and mtorc2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef]
- Alves, M.G.; Rato, L.; Carvalho, R.A.; Moreira, P.I.; Socorro, S.; Oliveira, P.F. Hormonal control of sertoli cell metabolism regulates spermatogenesis. Cell. Mol. Life Sci. 2013, 70, 777–793. [Google Scholar] [CrossRef]
- Oliveira, P.F.; Martins, A.D.; Moreira, A.C.; Cheng, C.Y.; Alves, M.G. The warburg effect revisited--lesson from the sertoli cell. Med. Res. Rev. 2015, 35, 126–151. [Google Scholar] [CrossRef]
- Martins, A.D.; Moreira, A.C.; Sa, R.; Monteiro, M.P.; Sousa, M.; Carvalho, R.A.; Silva, B.M.; Oliveira, P.F.; Alves, M.G. Leptin modulates human sertoli cells acetate production and glycolytic profile: A novel mechanism of obesity-induced male infertility? Biochim. Biophys. Acta 2015, 1852, 1824–1832. [Google Scholar] [CrossRef]
- Martins, A.D.; Sá, R.; Monteiro, M.P.; Barros, A.; Sousa, M.; Carvalho, R.A.; Silva, B.M.; Oliveira, P.F.; Alves, M.G. Ghrelin acts as energy status sensor of male reproduction by modulating sertoli cells glycolytic metabolism and mitochondrial bioenergetics. Mol. Cell. Endocrinol. 2016, 434, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.G.; Jesus, T.T.; Sousa, M.; Goldberg, E.; Silva, B.M.; Oliveira, P.F. Male fertility and obesity: Are ghrelin, leptin and glucagon-like peptide-1 pharmacologically relevant? Curr. Pharm. Des. 2016, 22, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.D.; Monteiro, M.P.; Silva, B.M.; Barros, A.; Sousa, M.; Carvalho, R.A.; Oliveira, P.F.; Alves, M.G. Metabolic dynamics of human sertoli cells are differentially modulated by physiological and pharmacological concentrations of glp-1. Toxicol. Appl. Pharmacol. 2019, 362, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.; McGowan, B. The glp-1 agonist, liraglutide, as a pharmacotherapy for obesity. Ther. Adv. Chronic Dis. 2016, 7, 92–107. [Google Scholar] [CrossRef] [PubMed]
 stimulation.
stimulation.  inhibition.
stimulation. inhibition.
inhibition.
stimulation. inhibition.
 stimulation. inhibition.
stimulation. inhibition.  downregulation/knockout.
downregulation/knockout.  upregulation.
stimulation. inhibition. downregulation/knockout. upregulation.
upregulation.
stimulation. inhibition. downregulation/knockout. upregulation.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, B.P.; Oliveira, P.F.; Alves, M.G. Molecular Mechanisms Controlled by mTOR in Male Reproductive System. Int. J. Mol. Sci. 2019, 20, 1633. https://doi.org/10.3390/ijms20071633
Moreira BP, Oliveira PF, Alves MG. Molecular Mechanisms Controlled by mTOR in Male Reproductive System. International Journal of Molecular Sciences. 2019; 20(7):1633. https://doi.org/10.3390/ijms20071633
Chicago/Turabian StyleMoreira, Bruno P., Pedro F. Oliveira, and Marco G. Alves. 2019. "Molecular Mechanisms Controlled by mTOR in Male Reproductive System" International Journal of Molecular Sciences 20, no. 7: 1633. https://doi.org/10.3390/ijms20071633
APA StyleMoreira, B. P., Oliveira, P. F., & Alves, M. G. (2019). Molecular Mechanisms Controlled by mTOR in Male Reproductive System. International Journal of Molecular Sciences, 20(7), 1633. https://doi.org/10.3390/ijms20071633