Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L.


Abstract
:1. Introduction
2. Results
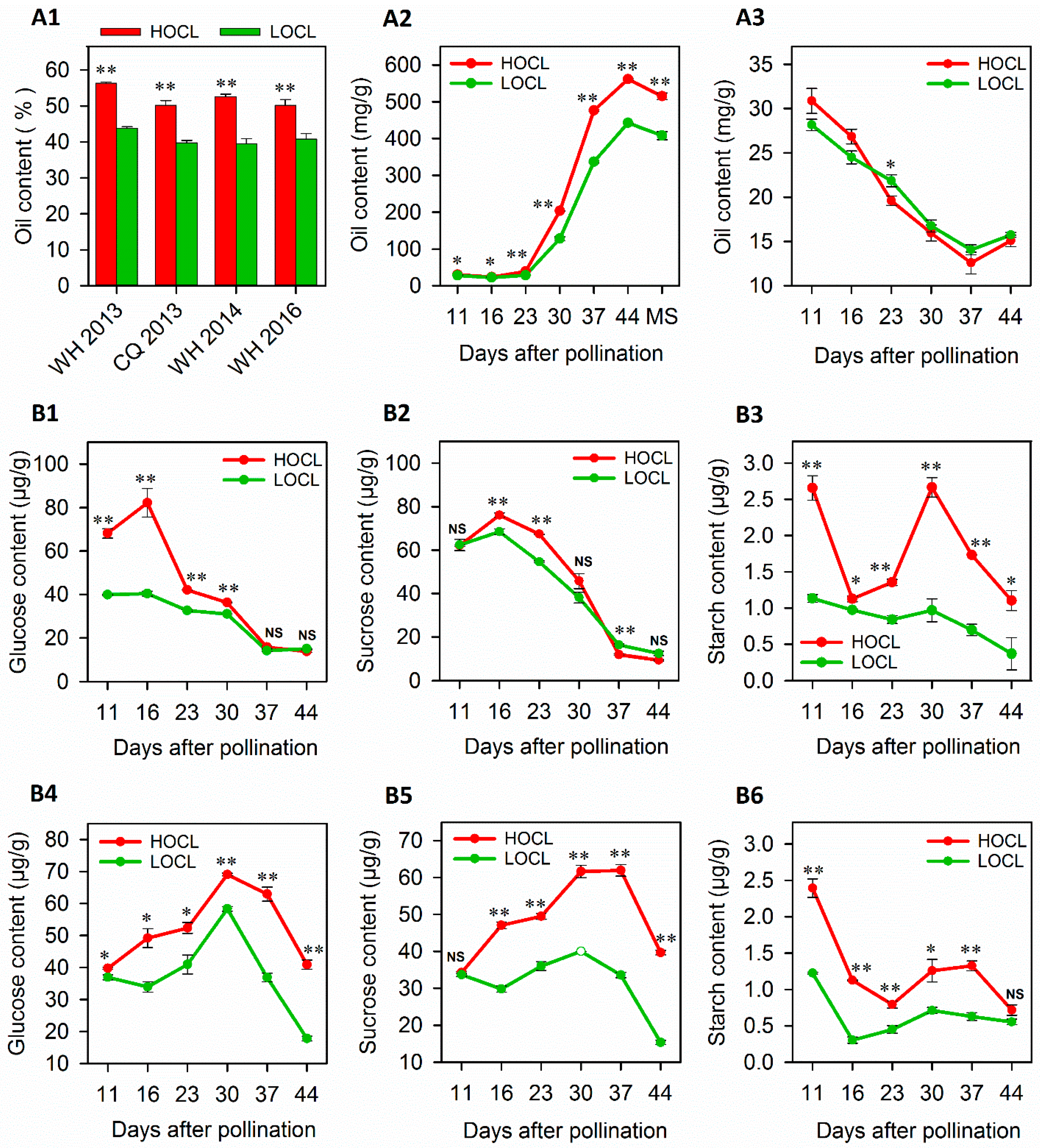
2.1. Dynamics of Oil Accumulation and Carbohydrates in HOC and LOC Lines
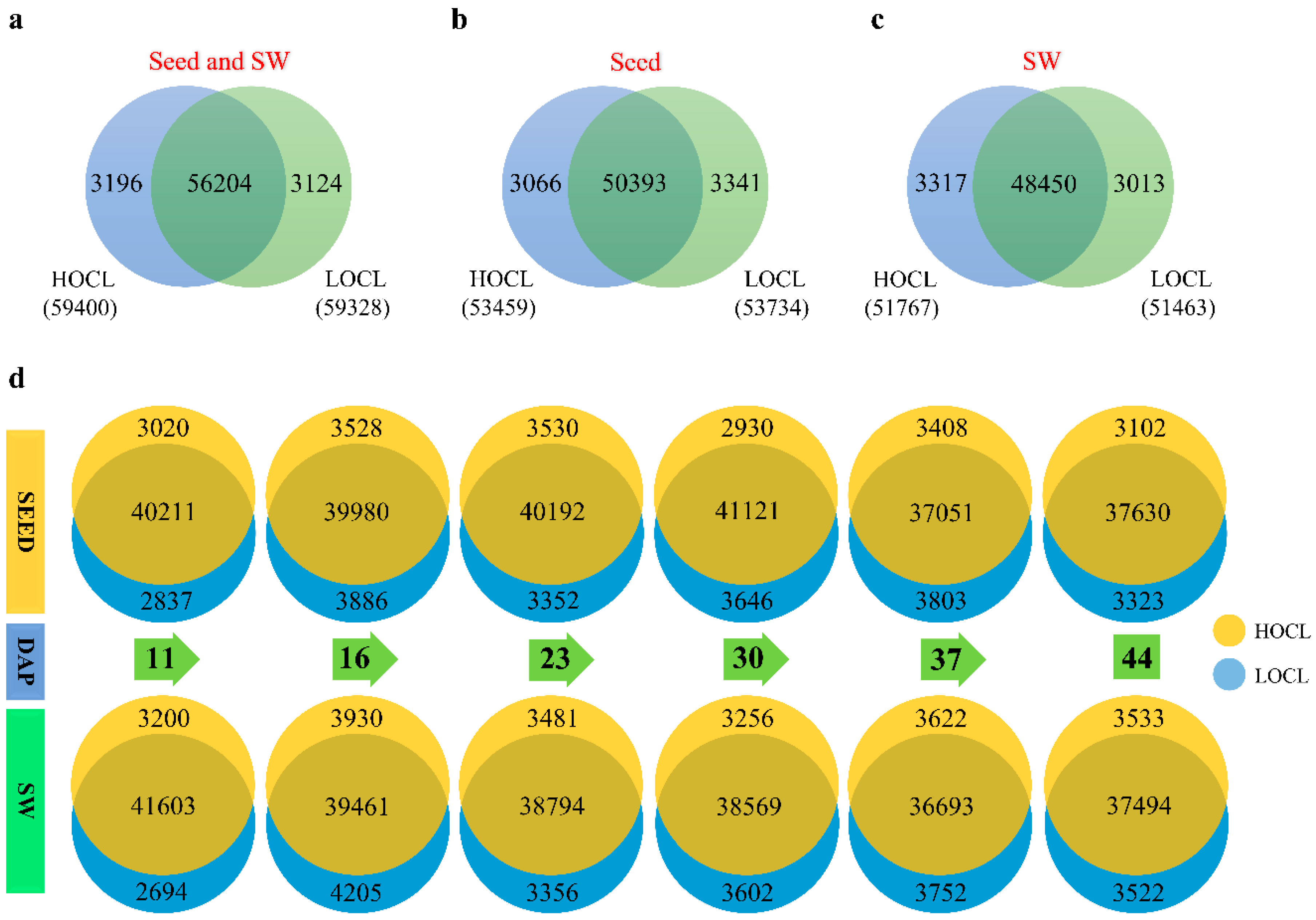
2.2. RNA Sequencing and Gene Expression Quantification Analysis
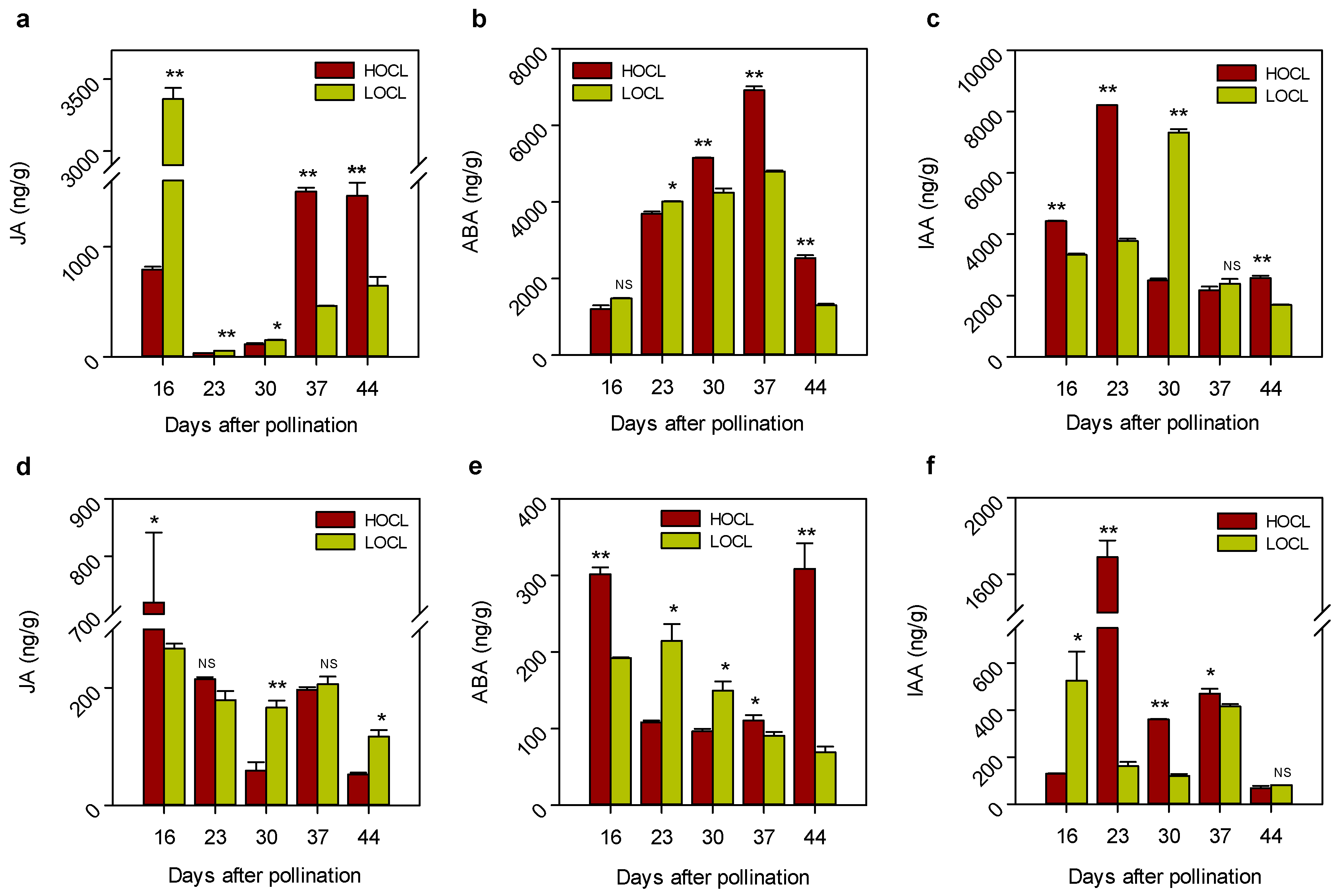
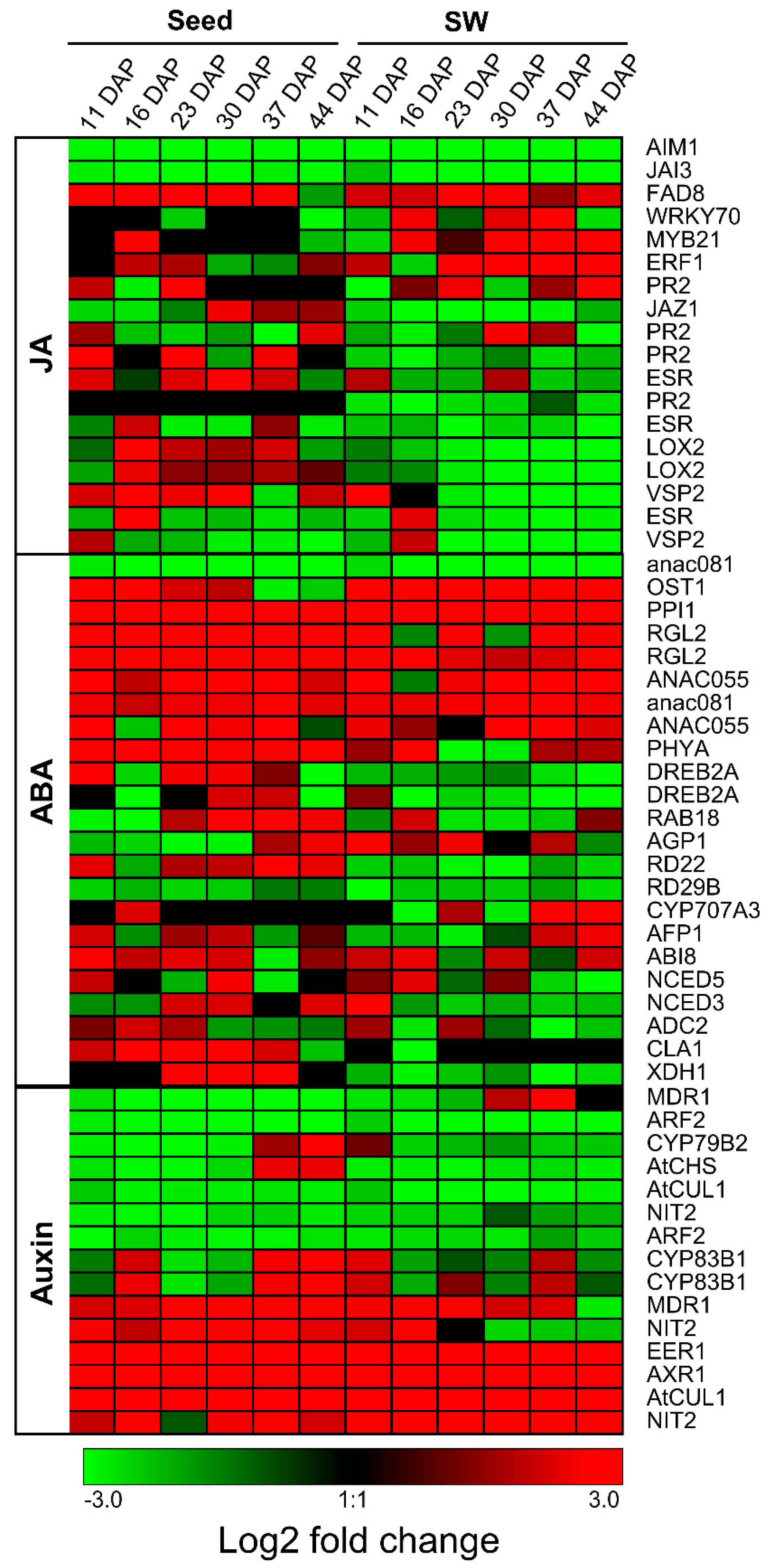
2.3. Expression Patterns of Genes Involved in Metabolic Pathways in High- and Low-Oil Content Lines
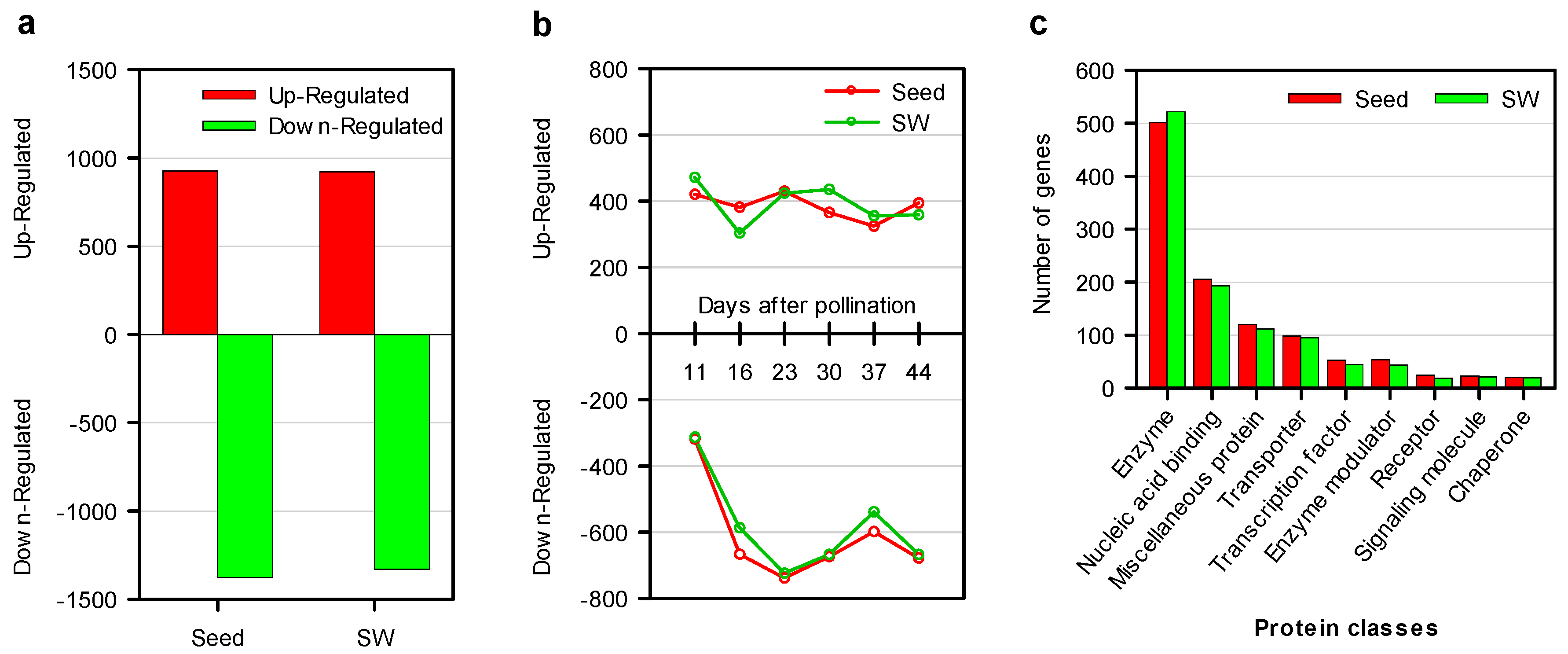
2.4. Comparative Dynamic Transcriptomics during Oil Accumulation between High- and Low-oil Lines
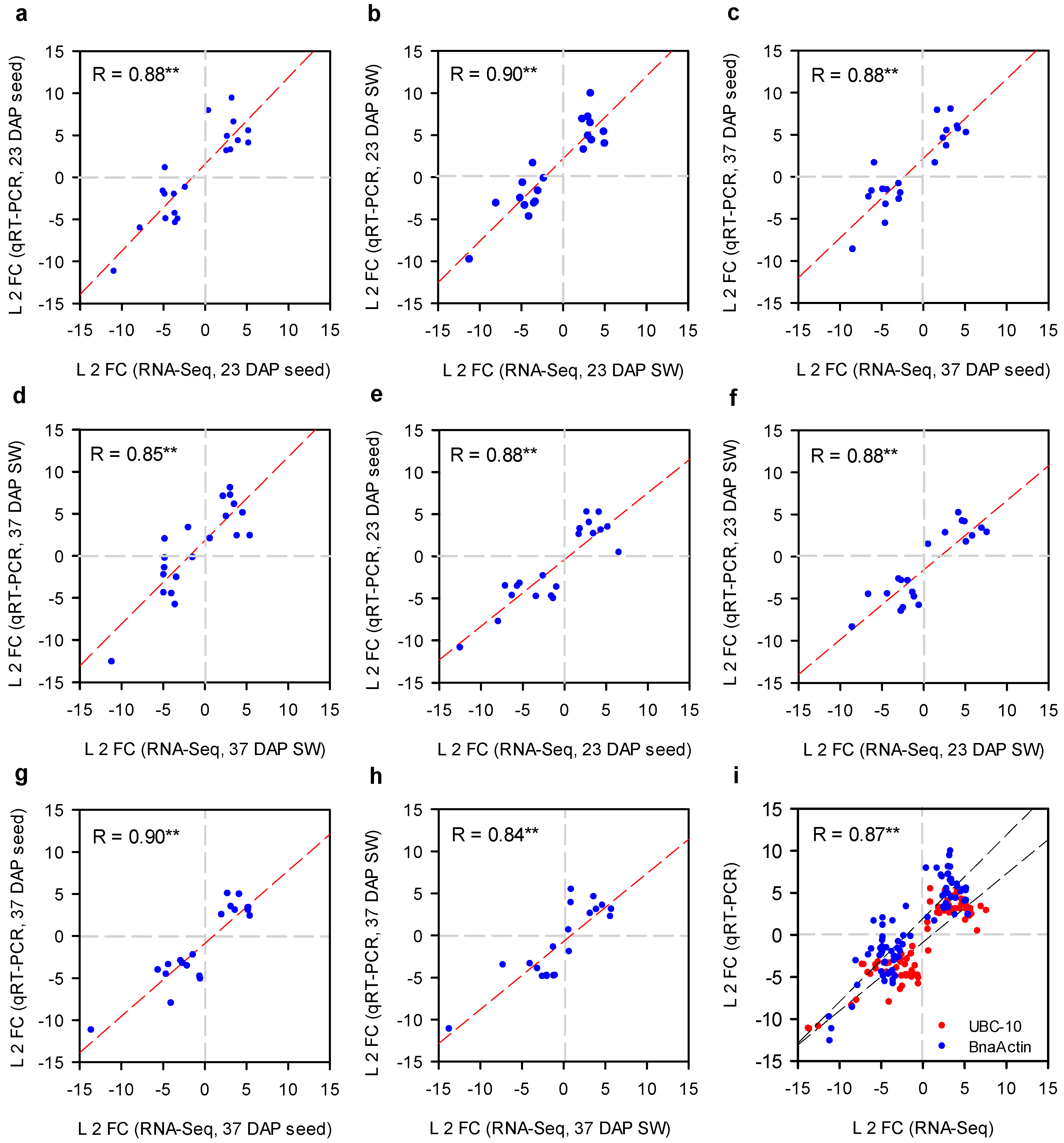
2.5. qRT-PCR Analysis of Selected DEGs to Validate RNA-Seq Results
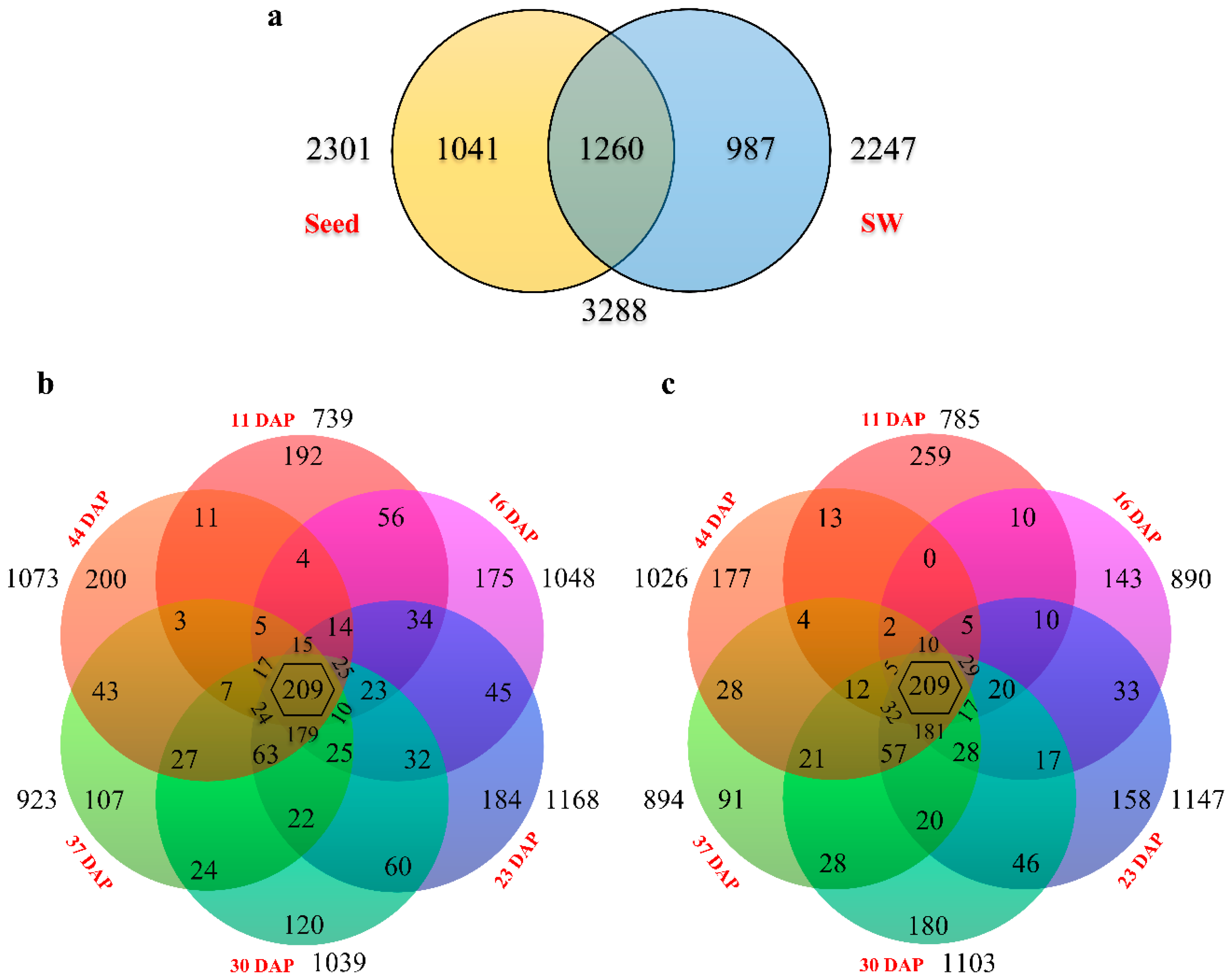
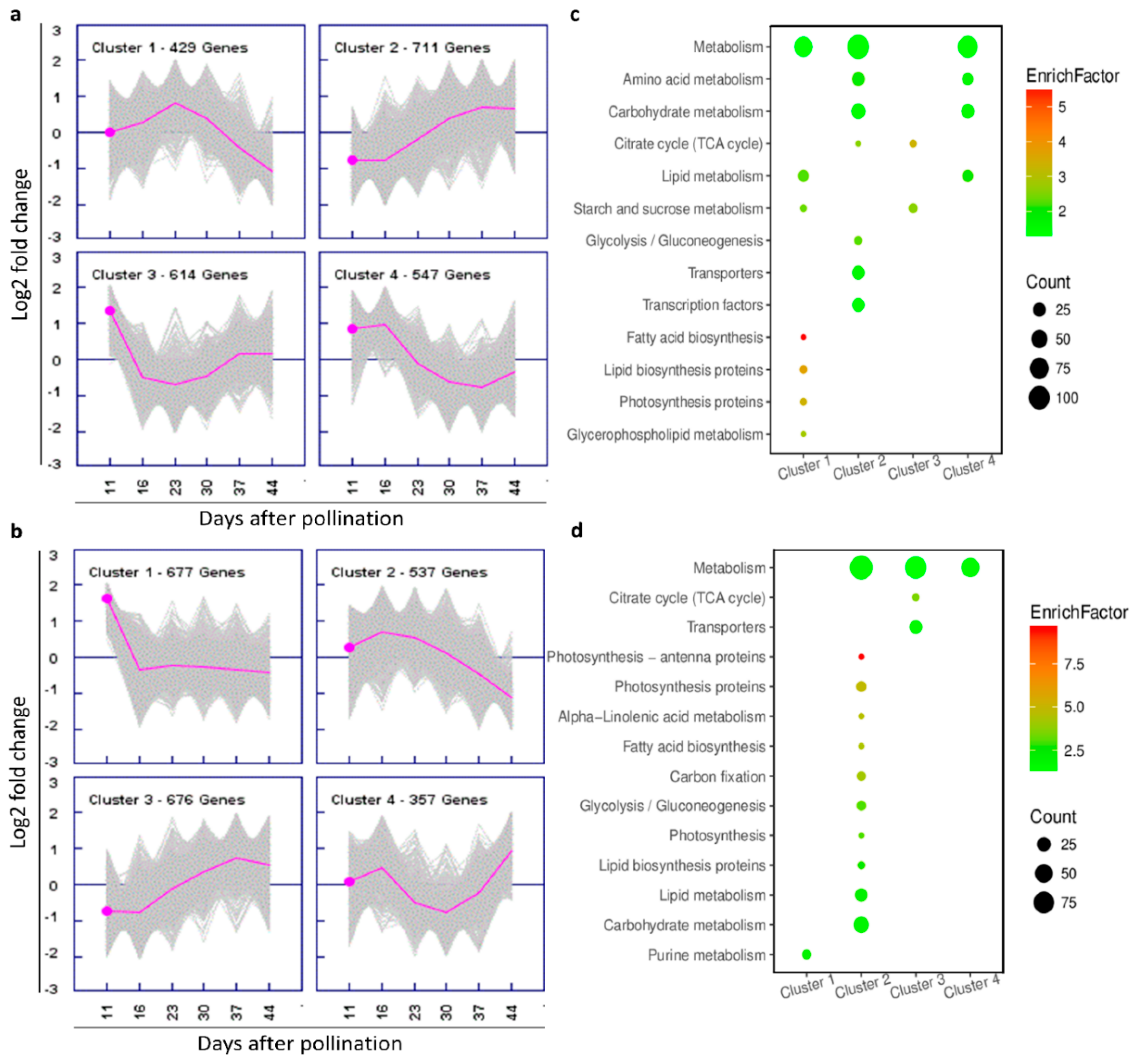
2.6. DEG Clustering
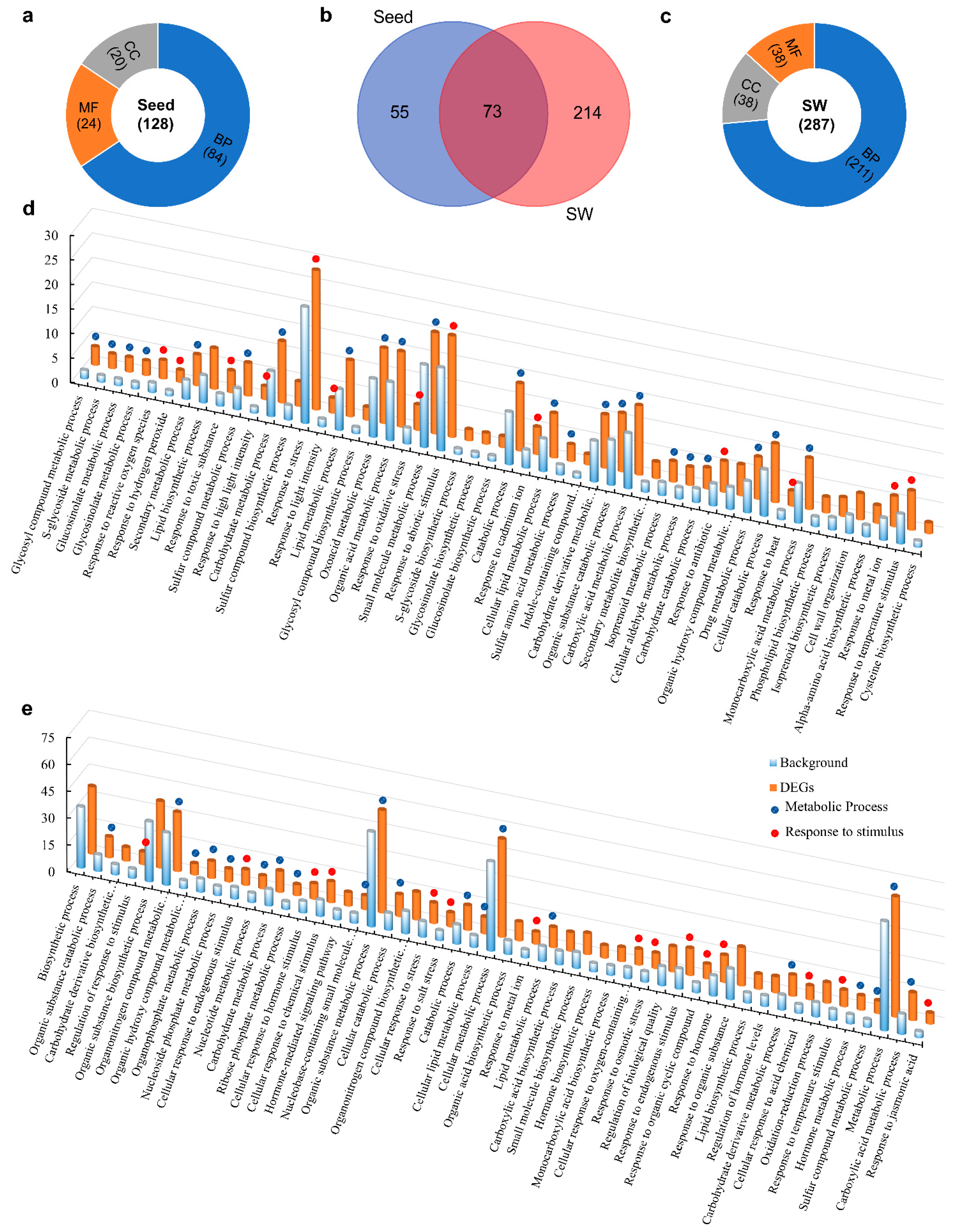
2.7. Functional Classifications of DEGs
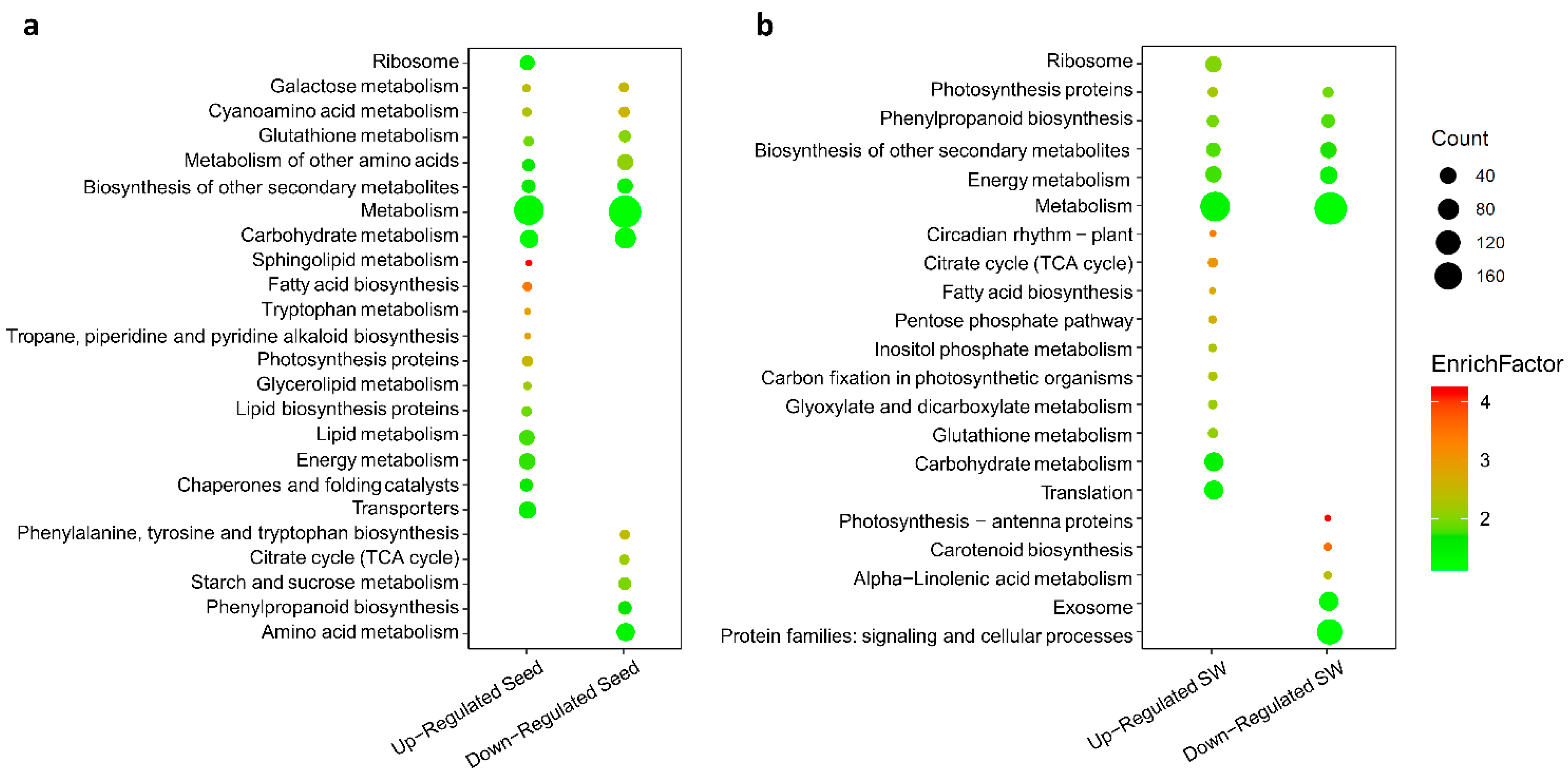
2.8. Identification of Up- and Down-regulated Pathways in Seed and SW
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. RNA Extraction, cDNA Library Construction, and Sequencing
4.3. Quality Control and Read Mapping to Reference Genome
4.4. Differential Gene Expression Quantification
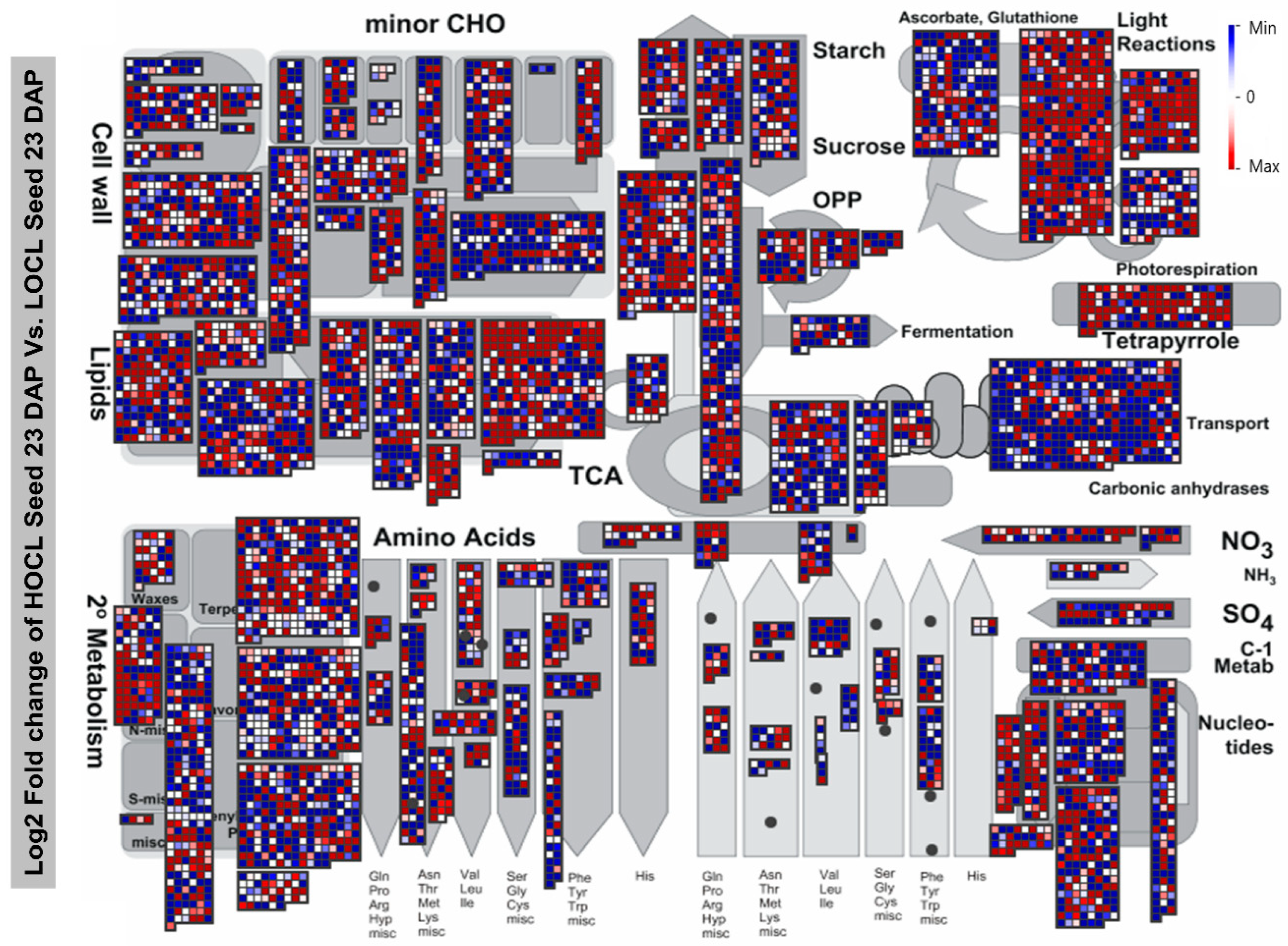
4.5. MapMan Visualization and Enrichment Analysis
4.6. Gene Ontology and Enrichment Analysis and KEGG Pathway Detection
4.7. Oil Content, Carbohydrates, and Hormone Quantification
4.8. Validation of RNA-Seq Data by qRT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALM | Acyl-lipid metabolism |
| DEGs | Differentially expressed genes |
| DAP | Days after pollination |
| FAs | Fatty acids |
| FDR | False discovery rate |
| GO | Gene ontology |
| HOCL | High oil content line |
| HPLC | High-performance liquid chromatograph |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LOCL | Low oil content line |
| OSR | Oilseed rape |
| RNA-Seq | RNA-sequencing |
| SOC | Seed oil content |
| TAG | Triacylglycerol |
| TFs | Transcription factors |
References
- Liu, S.; Fan, C.; Li, J.; Cai, G.; Yang, Q.; Wu, J.; Yi, X.; Zhang, C.; Zhou, Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 2016, 129, 1203–1215. [Google Scholar] [CrossRef]
- Lu, C.; Napier, J.A.; Clemente, T.E.; Cahoon, E.B. New frontiers in oilseed biotechnology: meeting the global demand for vegetable oils for food, feed, biofuel, and industrial applications. Curr. Opin. Biotech. 2011, 22, 252–259. [Google Scholar] [CrossRef]
- USDA-ERS, Oil Crops Yearbook [EB/OL]. Available online: https://www.ers.usda.gov/data-products/oil-crops-yearbook/oil-crops-yearbook/#Canola%20Seed%20and%20Canola%20Seed%20Products (accessed on 19 April 2019).
- Troncoso-Ponce, M.A.; Kilaru, A.; Cao, X.; Durrett, T.P.; Fan, J.; Jensen, J.K.; Thrower, N.A.; Pauly, M.; Wilkerson, C.; Ohlrogge, J.B. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011, 68, 1014–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Z. Strategy for rapeseed genetic improvement in China in the coming fifteen years. Chin. J. Oil Crop Sci. 2004, 26, 98–101. [Google Scholar]
- Hajduch, M.; Casteel, J.E.; Hurrelmeyer, K.E.; Song, Z.; Agrawal, G.K.; Thelen, J.J. Proteomic analysis of seed filling in Brassica napus. Developmental characterization of metabolic isozymes using high-resolution two-dimensional gel electrophoresis. Plant Physiol. 2006, 141, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H. Oil bodies and oleosins in seeds. Annu. Rev. Plant Biol. 1992, 43, 177–200. [Google Scholar] [CrossRef]
- Goldberg, R.B.; De Paiva, G.; Yadegari, R. Plant embryogenesis: zygote to seed. Science 1994, 266, 605–614. [Google Scholar] [CrossRef]
- Murphy, D.J.; Cummins, I.; Kang, A.S. Synthesis of the major oil-body membrane protein in developing rapeseed (Brassica napus) embryos. Integration with storage-lipid and storage-protein synthesis and implications for the mechanism of oil-body formation. Biochem. J. 1989, 258, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 2010, 49, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Wu, G.-Z.; Ye, R.; Lin, W.-H.; Shi, Q.-M.; Xue, L.-J.; Xu, X.-D.; Li, Y.; Du, Y.-G.; Xue, H.-W. Global analysis of gene expression profiles in Brassica napus developing seeds reveals a conserved lipid metabolism regulation with Arabidopsis thaliana. Mol. Plant 2009, 2, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Chen, Z.-H.; Zhang, Y.; Yu, H.; Lin, B.; Zhang, D. Chlorophyll and carbohydrate metabolism in developing silique and seed are prerequisite to seed oil content of Brassica napus L. Bot. Stud. 2014, 55, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Q.; Fan, C.; Zhao, X.; Wang, X.; Zhou, Y. Transcriptomic basis of functional difference and coordination between seeds and the silique wall of Brassica napus during the seed-filling stage. Plant Sci. 2015, 233, 186–199. [Google Scholar] [CrossRef]
- Sessions, R.A.; Zambryski, P.C. Arabidopsis gynoecium structure in the wild and in ettin mutants. Development 1995, 121, 1519–1532. [Google Scholar] [PubMed]
- King, S.P.; Badger, M.R.; Furbank, R.T. CO2 refixation characteristics of developing canola seeds and silique wall. Funct. Plant Biol. 1998, 25, 377–386. [Google Scholar] [CrossRef]
- Ma, Q.; Behboudian, M.; Turner, N.C.; Palta, J.A. Gas exchange by pods and subtending leaves and internal recycling of CO2 by pods of chickpea (Cicer arietinum L.) subjected to water deficits. J. Exp. Bot. 2001, 52, 123–131. [Google Scholar] [CrossRef]
- Furbank, R.T.; White, R.J.; Palta, A.; Turner, N.C. Internal recycling of respiratory CO2 in pods of chickpea (Cicer arietinum L.): the role of pod wall, seed coat, and embryo. J. Exp. Bot. 2004, 55, 1687–1696. [Google Scholar] [CrossRef]
- Schwender, J.; Goffman, F.; Ohlrogge, J.B.; Shachar-Hill, Y. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature 2004, 432, 779–782. [Google Scholar] [CrossRef]
- Beisson, F.; Koo, A.J.; Ruuska, S.; Schwender, J.; Pollard, M.; Thelen, J.J.; Paddock, T.; Salas, J.J.; Savage, L.; Milcamps, A. Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol. 2003, 132, 681–697. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Singh, R.; Ong-Abdullah, M.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.E.; Chan, K.-L.; Halim, M.A.; et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 2013, 500, 335–339. [Google Scholar] [CrossRef]
- Wang, J.; Singh, S.K.; Du, C.; Li, C.; Fan, J.; Pattanaik, S.; Yuan, L. Comparative Transcriptomic analysis of two Brassica napus near-Isogenic lines reveals a network of genes that influences seed oil accumulation. Front. Plant Sci. 2016, 7, 1498. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, G.; Cao, Y.; Wu, Y.; Xiao, L.; Li, X.; Lu, C. Breeding response of transcript profiling in developing seeds of Brassica napus. BMC Mol. Biol. 2009, 10, 1–17. [Google Scholar] [CrossRef]
- Deng, W.; Yan, F.; Zhang, X.; Tang, Y.; Yuan, Y. Transcriptional profiling of canola developing embryo and identification of the important roles of BnDof5. 6 in embryo development and fatty acids synthesis. Plant Cell Physiol. 2015, 56, 1624–1640. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, R.; Friedt, W. Molecular markers in Brassica oilseed breeding: current status and future possibilities. Plant Breeding 2004, 123, 1–8. [Google Scholar] [CrossRef]
- Parkin, I.A.; Gulden, S.M.; Sharpe, A.G.; Lukens, L.; Trick, M.; Osborn, T.C.; Lydiate, D.J. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 2005, 171, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Lardizabal, K.; Effertz, R.; Levering, C.; Mai, J.; Pedroso, M.; Jury, T.; Aasen, E.; Gruys, K.; Bennett, K. Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol. 2008, 148, 89–96. [Google Scholar] [CrossRef]
- Wan, H.; Cui, Y.; Ding, Y.; Mei, J.; Dong, H.; Zhang, W.; Wu, S.; Liang, Y.; Zhang, C.; Li, J. Time-series analyses of transcriptomes and proteomes reveal molecular networks underlying oil accumulation in Canola. Front. Plant Sci. 2017, 7, 1–17. [Google Scholar] [CrossRef]
- Hua, W.; Li, R.J.; Zhan, G.M.; Liu, J.; Li, J.; Wang, X.F.; Liu, G.H.; Wang, H.Z. Maternal control of seed oil content in Brassica napus: the role of silique wall photosynthesis. Plant J. 2012, 69, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-L.; Zhang, M.-L.; Ma, G.-J.; Wu, H.; Wu, X.-M.; Ren, F.; Li, X.-B. Transcriptome profiling analysis reveals the role of silique in controlling seed oil content in Brassica napus. PLoS ONE 2017, 12, e0179027. [Google Scholar] [CrossRef]
- Rossato, L.L.P.; Ourry, A. Nitrogen storage and remobilization in Brassica napus L. during the growth cycle: nitrogen fluxes within the plant and changes in soluble protein patterns. J. Exp. Bot. 2001, 52, 1623–1655. [Google Scholar] [CrossRef]
- Dubousset, L.; Etienne, P.; Avice, J. Is the remobilization of S and N reserves for seed filling of winter oilseed rape modulated by sulphate restrictions occurring at different growth stages? J. Exp. Bot. 2010, 61, 4313–4324. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.; Onge, R.P.S.; Hartman, J.L.; Giaever, G.; Roth, F.P. Defining genetic interaction. Proc. Nat. Acad. Sci. USA 2008, 105, 3461–3466. [Google Scholar] [CrossRef] [Green Version]
- Shah, J. Lipids, lipases, and lipid-modifying enzymes in plant disease resistance. Annu. Rev. Phytopathol. 2005, 43, 229–260. [Google Scholar] [CrossRef]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: unraveling the signaling networks. Front. Plant Sci. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Higashi, Y.; Okazaki, Y.; Myouga, F.; Shinozaki, K.; Saito, K. Landscape of the lipidome and transcriptome under heat stress in Arabidopsis thaliana. Sci. Rep. 2015, 5, 10533. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Ufer, G.; Bartels, D. Lipid signalling in plant responses to abiotic stress. Plant Cell Environ. 2016, 39, 1029–1048. [Google Scholar] [CrossRef]
- Fallahi, H.; Scofield, G.N.; Badger, M.R.; Chow, W.S.; Furbank, R.T.; Ruan, Y.-L. Localization of sucrose synthase in developing seed and siliques of Arabidopsis thaliana reveals diverse roles for SUS during development. J. Exp. Bot. 2008, 59, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Andriotis, V.M.; Pike, M.J.; Kular, B.; Rawsthorne, S.; Smith, A.M. Starch turnover in developing oilseed embryos. New Phytol. 2010, 187, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Shen, Y.; Hao, J.; Wu, J.; Ke, L.; Wu, C.; Huang, K.; Luo, B.; Xu, M.; Cheng, X. Acyl-CoA N-acyltransferase influences fertility by regulating lipid metabolism and jasmonic acid biogenesis in cotton. Sci. Rep. 2015, 5, 11790. [Google Scholar] [CrossRef] [Green Version]
- Kachroo, A.; Lapchyk, L.; Fukushige, H.; Hildebrand, D.; Klessig, D.; Kachroo, P. Plastidial fatty acid signaling modulates salicylic acid–and jasmonic acid–mediated defense pathways in the Arabidopsis ssi2 mutant. Plant Cell 2003, 15, 2952–2965. [Google Scholar] [CrossRef]
- Nandi, A.; Krothapalli, K.; Buseman, C.M.; Li, M.; Welti, R.; Enyedi, A.; Shah, J. Arabidopsis sfd mutants affect plastidic lipid composition and suppress dwarfing, cell death, and the enhanced disease resistance phenotypes resulting from the deficiency of a fatty acid desaturase. Plant Cell 2003, 15, 2383–2398. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Abrams, G.D.; Barton, D.L.; Taylor, D.C.; Pomeroy, M.K.; Abrams, S.R. Induction of lipid and oleosin biosynthesis by (+)-abscisic acid and its metabolites in microspore-derived embryos of Brassica napus L. cv Reston (biological responses in the presence of 8 [prime]-hydroxyabscisic acid). Plant Physiol. 1995, 108, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.S.; Taylor, D.C.; Giblin, M.; Ferrie, A.M.; Ambrose, S.J.; Ross, A.R.; Nelson, K.M.; Zaharia, L.I.; Sharma, N.; Anderson, M. Hormonal regulation of oil accumulation in Brassica seeds: metabolism and biological activity of ABA, 7′-, 8′-and 9′-hydroxy ABA in microspore derived embryos of B. napus. Phytochemistry 2008, 69, 2678–2688. [Google Scholar] [CrossRef]
- Tan, H.; Yang, X.; Zhang, F.; Zheng, X.; Qu, C.; Mu, J.; Fu, F.; Li, J.; Guan, R.; Zhang, H. Enhanced seed oil production in canola by conditional expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant Physiol. 2011, 156, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Allen, W.B.; Zheng, P.; Li, C.; Glassman, K.; Ranch, J.; Nubel, D.; Tarczynski, M.C. Expression of ZmLEC1 and ZmWRI1 increases seed oil production in maize. Plant Physiol. 2010, 153, 980–987. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, R.W.; Siol, M.; Barrett, S.C. De novo sequence assembly and characterization of the floral transcriptome in cross-and self-fertilizing plants. BMC Genom. 2011, 12, 1–13. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Bahn, S.C.; Qu, G.; Qin, H.; Hong, Y.; Xu, Q.; Zhou, Y.; Hong, Y.; Wang, X. Increased expression of phospholipase Dα1 in guard cells decreases water loss with improved seed production under drought in Brassica napus. Plant Biotechnol. J. 2013, 11, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.; Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Chow, P.S.; Landhäusser, S.M. A method for routine measurements of total sugar and starch content in woody plant tissues. Tree Physiol. 2004, 24, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Welti, R.; Wang, X. Simultaneous quantification of major phytohormones and related compounds in crude plant extracts by liquid chromatography–electrospray tandem mass spectrometry. Phytochemistry 2008, 69, 1773–1781. [Google Scholar] [PubMed]
- Liu, H.; Li, X.; Xiao, J.; Wang, S. A convenient method for simultaneous quantification of multiple phytohormones and metabolites: application in study of rice-bacterium interaction. Plant Methods 2012, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, Q.; Yang, Q.; Liu, H.; Li, Q.; Yi, X.; Cheng, Y.; Guo, L.; Fan, C.; Zhou, Y. Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 2016, 6, e19007. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shao, J.; Tang, S.; Shen, Q.; Wang, T.; Chen, W.; Hong, Y. Wrinkled1 accelerates flowering and regulates lipid homeostasis between oil accumulation and membrane lipid anabolism in Brassica napus. Front. Plant Sci. 2016, 6, 1270. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bin | Assignment a | All b | HOCL-Up | LOCL-Up | p-Value |
|---|---|---|---|---|---|
| 1 | Photosynthesis | 556 (0.89) | 353 (1.18) | 193 (0.64) | 2.51 × 10−13 ** |
| 2 | Major carbohydrates metabolism | 268 (0.43) | 127 (0.42) | 134 (0.44) | 0.77 |
| 3 | Minor carbohydrates metabolism | 318 (0.51) | 150 (0.50) | 158 (0.52) | 0.60 |
| 4 | Glycolysis | 233 (0.37) | 103 (0.34) | 125 (0.41) | 0.07 |
| 5 | Fermentation | 42 (0.07) | 14 (0.05) | 28 (0.09) | 1.8 × 10−3 ** |
| 6 | Gluconeogenesis/glyoxylate cycle | 44 (0.07) | 19 (0.06) | 20 (0.07) | 0.76 |
| 7 | Oxidative PP cycle | 86 (0.14) | 48 (0.16) | 38 (0.13) | 0.35 |
| 8 | TCA/organic acid transformation | 235 (0.38) | 83 (0.28) | 138 (0.46) | 1.0 × 10−3 ** |
| 9 | Mitochondrial transport/ATP synthesis | 366 (0.59) | 103 (0.34) | 254 (0.84) | 6.0 × 10−16 ** |
| 10 | Cell wall (Degradation) | 1178 (1.88) | 511 (1.70) | 574 (1.89) | 0.02 * |
| 11 | Lipid metabolism (FA desaturation) | 1133 (1.81) | 556 (1.85) | 524 (1.73) | 4.6 × 10−2 * |
| 12 | N-metabolism | 80 (0.13) | 40 (0.13) | 40 (0.13) | 0.81 |
| 13 | Amino acid metabolism | 800 (1.28) | 345 (1.15) | 432 (1.43) | 2.7 × 10−3 ** |
| 14 | S-assimilation | 42 (0.07) | 16 (0.05) | 25 (0.08) | 0.18 |
| 15 | Metal handling | 187 (0.30) | 77 (0.26) | 97 (0.32) | 0.17 |
| 16 | Secondary metabolism | 1122 (1.79) | 469 (1.56) | 529 (1.75) | 0.06 |
| 17 | Hormone metabolism | 1524 (2.44) | 671 (2.24) | 657 (2.17) | 0.25 |
| 18 | Co-factor and vitamin metabolism | 216 (0.35) | 87 (0.29) | 126 (0.42) | 0.01* |
| 19 | Tetrapyrrole synthesis | 122 (0.20) | 79 (0.26) | 39 (0.13) | 9.2 × 10−6 ** |
| 20 | Stress (biotic and abiotic) | 2379 (3.80) | 978 (3.26) | 1114 (3.67) | 8.6 × 10−4 ** |
| 21 | Redox regulation (Thioredoxin) | 604 (0.97) | 227 (0.76) | 329 (1.09) | 4.18 × 10−5 ** |
| 22 | Polyamine metabolism | 55 (0.09) | 30 (0.10) | 23 (0.08) | 0.06 |
| 23 | Nucleotide metabolism | 482 (0.77) | 221 (0.74) | 248 (0.82) | 0.14 |
| 24 | Biodegradation of Xenobiotic | 78 (0.12) | 26 (0.09) | 49 (0.16) | 1.6 × 10−3 ** |
| 25 | C1-metabolism | 101 (0.16) | 35 (0.12) | 63 (0.21) | 5.8 × 10−3 ** |
| 26 | Miscellaneous enzyme families | 3319 (5.31) | 1431 (4.77) | 1520 (5.01) | 0.20 |
| 27 | RNA | 7875 (12.6) | 3950 (13.1) | 3434 (11.3) | 2.4 × 10−12 ** |
| 28 | DNA (Synthesis repair) | 1127 (1.80 | 641 (2.14) | 466 (1.54) | 1.4 × 10−9 ** |
| 29 | Protein | 9862 (15.7) | 4909 (16.3) | 4652 (15.3) | 4.6 × 10−3 ** |
| 30 | Signaling | 3222 (5.15) | 1508 (5.03) | 1470 (4.85) | 0.94 |
| 31 | Cell (Vesicle transport) | 2150 (3.44) | 1036 (3.46) | 1073 (3.54) | 0.92 |
| 32 | Micro RNA, natural antisense etc. | 3 (0.004) | 0 (0) | 3 (0.01) | 0.47 |
| 33 | Development | 2027 (3.24) | 958 (3.20) | 885 (2.92) | 1.9 × 10−2 * |
| 34 | Transport (Metabolite transporters) | 2670 (4.27) | 1306 (4.36) | 1235 (4.07) | 5.1 × 10−7 ** |
| 35 | Not assigned | 20,586 (32.92) | 8876 (29.60) | 9618 (31.7) | 4.8 × 10−8 ** |
| Total | 62,524 | 29,983 | 30,313 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahid, M.; Cai, G.; Zu, F.; Zhao, Q.; Qasim, M.U.; Hong, Y.; Fan, C.; Zhou, Y. Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L. Int. J. Mol. Sci. 2019, 20, 1982. https://doi.org/10.3390/ijms20081982
Shahid M, Cai G, Zu F, Zhao Q, Qasim MU, Hong Y, Fan C, Zhou Y. Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L. International Journal of Molecular Sciences. 2019; 20(8):1982. https://doi.org/10.3390/ijms20081982
Chicago/Turabian StyleShahid, Muhammad, Guangqin Cai, Feng Zu, Qing Zhao, Muhammad Uzair Qasim, Yueyun Hong, Chuchuan Fan, and Yongming Zhou. 2019. "Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L." International Journal of Molecular Sciences 20, no. 8: 1982. https://doi.org/10.3390/ijms20081982
APA StyleShahid, M., Cai, G., Zu, F., Zhao, Q., Qasim, M. U., Hong, Y., Fan, C., & Zhou, Y. (2019). Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L. International Journal of Molecular Sciences, 20(8), 1982. https://doi.org/10.3390/ijms20081982