UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. UBA6
2.1. UBA6 with Neuronal Diseases
2.2. UBA6 with Cancer Development
2.3. UBA6 with Meiosis Initiation
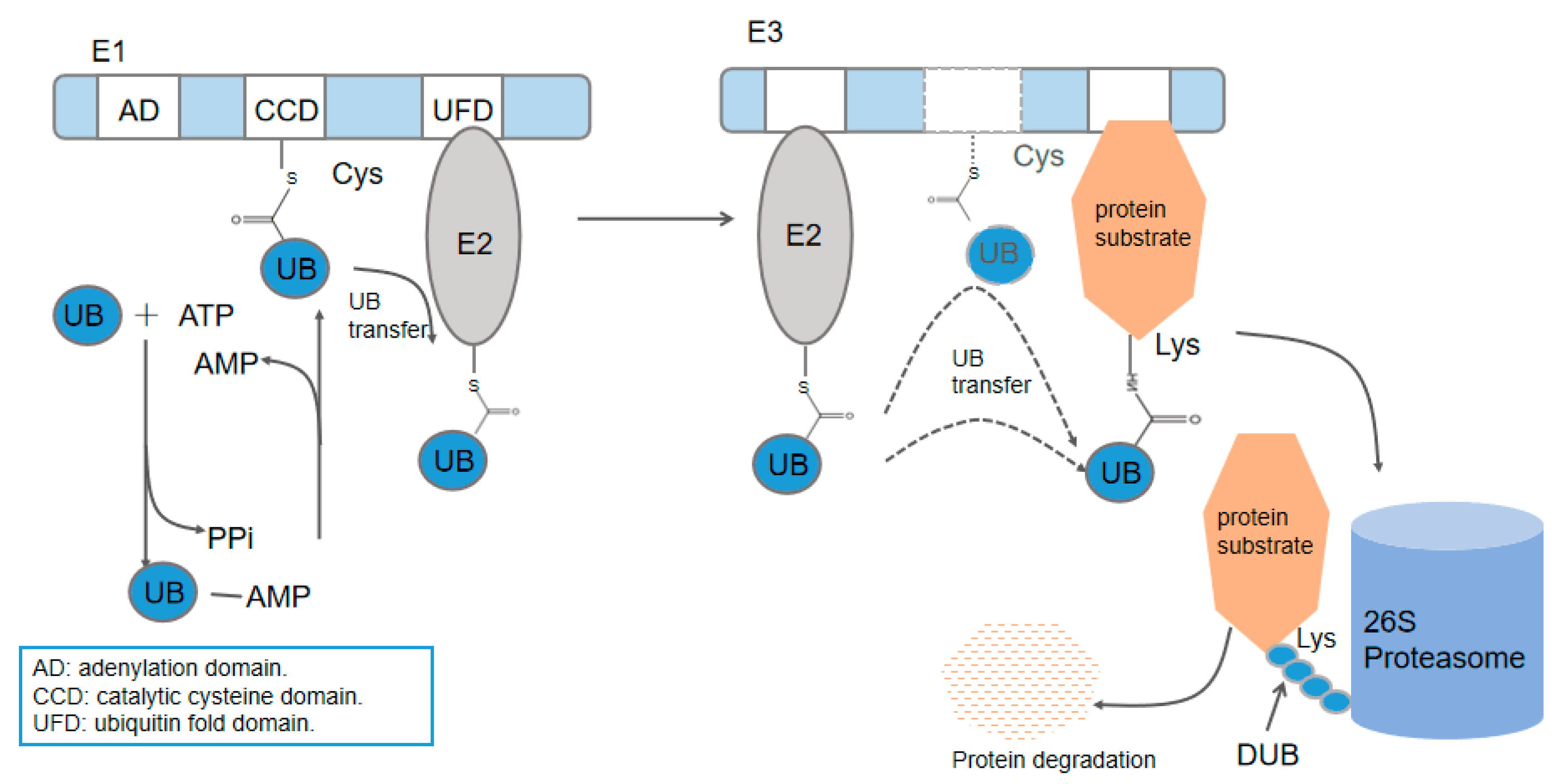
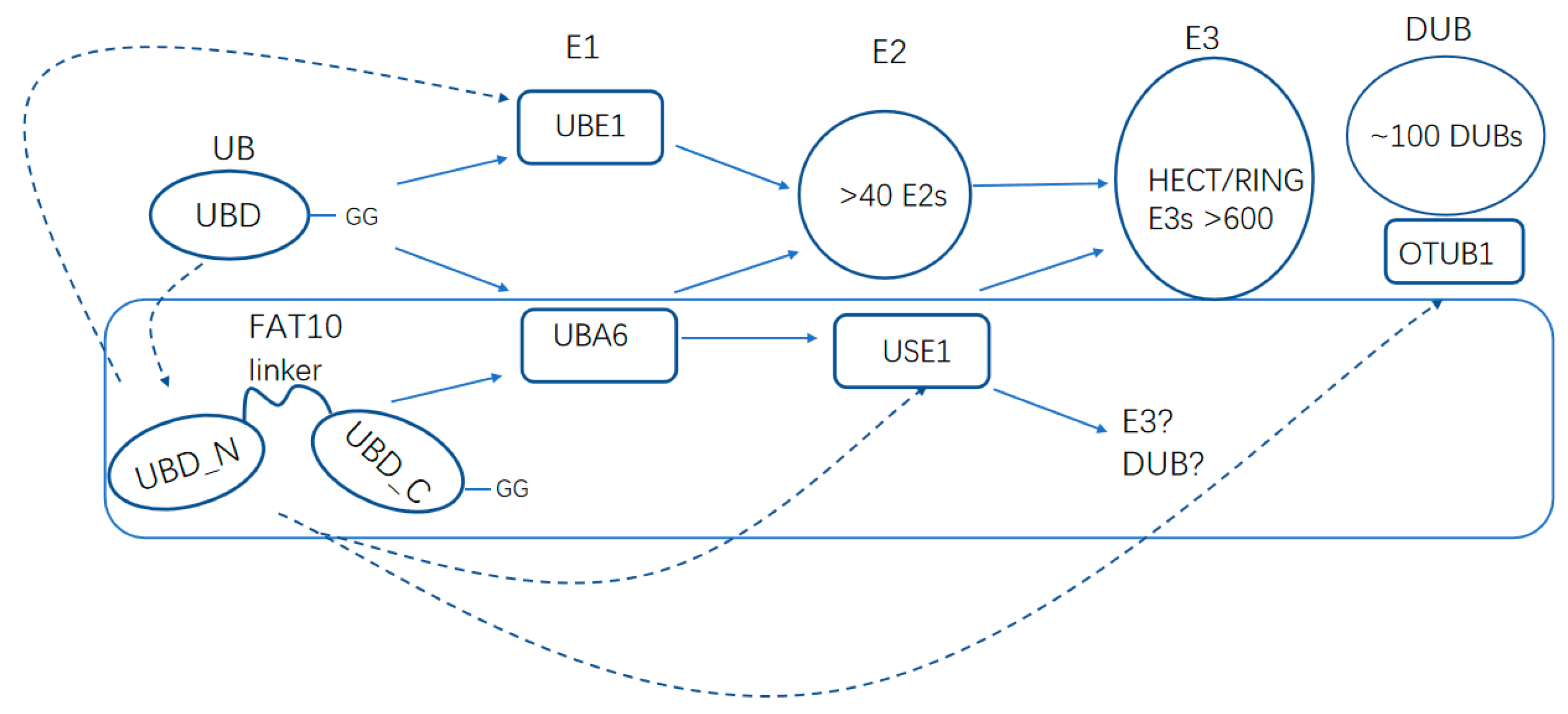
3. The UBA6–Ubiquitin Cascades
3.1. Spatial Differences of the UBA6 and UBE1 Cascades
3.2. Identified Ubiquitination Cascades
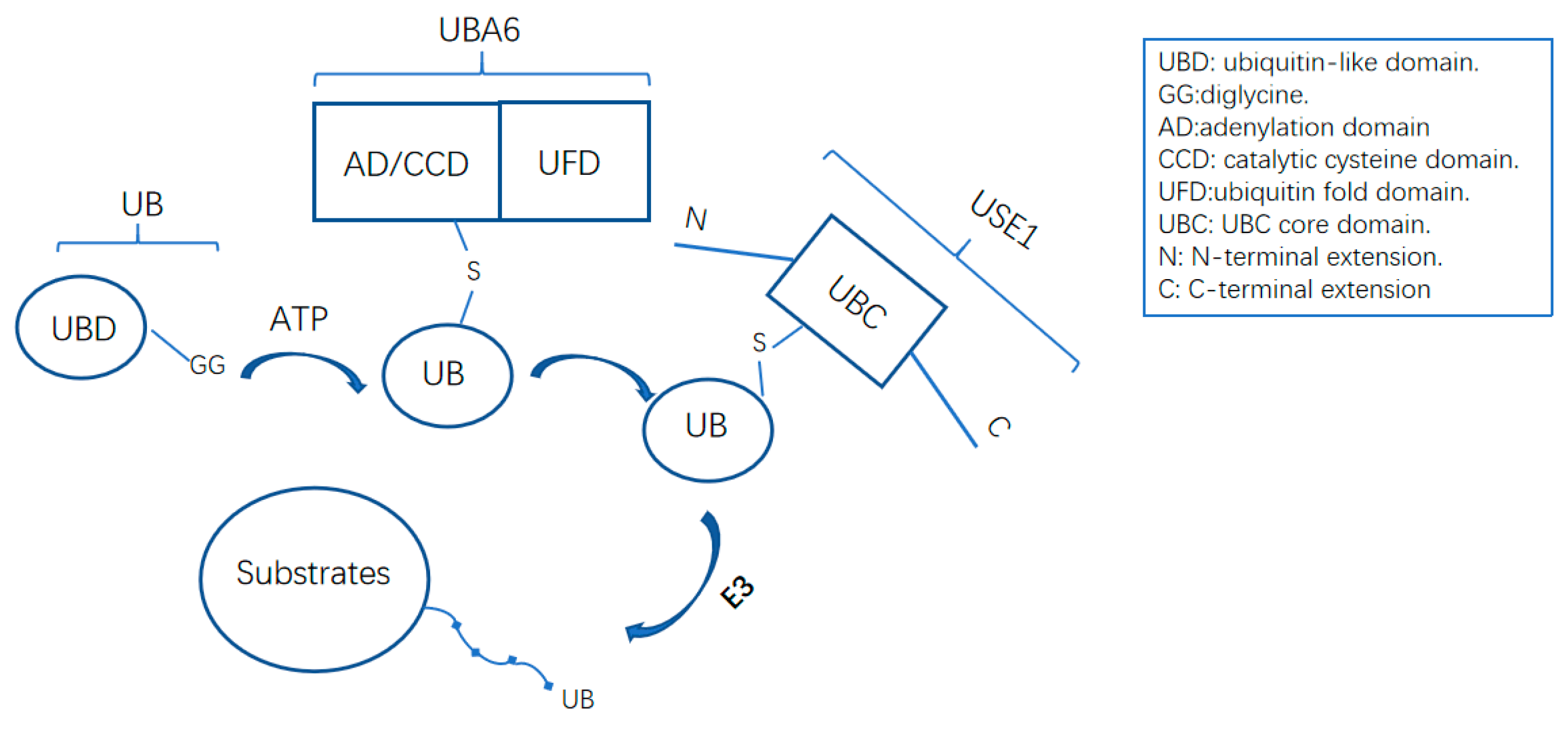
4. The UBA6–FAT10 Cascades
4.1. The FAT10 Structure
4.2. Regulation of the FAT10 Cascade
5. Candidates of FAT10ylation Cascades
Does FAT10 Facilitate Protein Degradation or Not?
6. Comparison and Interplay between FAT10 and Ubiquitin
6.1. Comparison
6.2. Interplay
7. USE1
7.1. USE1 with Cardiovascular Diseases
7.2. USE1 with Human Lung Cancer
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AIPL1 | Aryl hydrocarbon receptor interacting protein-like 1 |
| APC/C | Anaphase-promoting complex/cyclosome |
| ASD | Autism spectrum disorder |
| Atg8 | Autophagy-related protein 8 |
| Atg12 | Autophagy related protein 12 |
| CAD | Coronary artery disease |
| CUGBP1 | CUG triplet repeat binding protein 1 |
| DUB | Deubiquitinase |
| eEF1A1 | eukaryotic translation elongation factor 1A1 |
| EMT | Epithelial–mesenchymal transition |
| EZR | Ezrin |
| FAT10 | Human leukocyte antigen (HLA)-F-adjacent transcript 10 |
| GIP | Gastric inhibitory peptide |
| GWAS | Genome-wide association studies |
| HCC | Hepatocellular carcinoma |
| HDAC6 | Histone deacetylase 6 |
| HECT | Homologous to E6AP C terminus |
| ID | Intellectual disorder |
| ISG15 | Interferon-stimulated gene 15 |
| JNK1/2 | c-jun-N-terminal kinase 1/2 |
| LMO2 | LIM domain only 2 |
| MAD2 | Mitotic arrest-deficient 2 |
| MDB | Mallory–Denk body |
| MI | Myocardial infarction |
| NEDD8 | Neural precursor cell expressed, developmentally down-regulated 8 |
| NUB1L | NEDD8 ultimate buster-1L |
| OTUB1 | OTU deubiquitinase, ubiquitin aldehyde binding 1 |
| OUT | Orthogonal ubiquitin transfer |
| RGS | Regulator of G-protein signaling |
| SUMO1-3 | Small ubiquitin-like modifiers 1–3 |
| UB | Ubiquitin |
| UBA6 | Ubiquitin-like modifier-activating enzyme 6 |
| UBC | Ubiquitin conjugating enzyme; UBC core domain |
| UBE1 | Ubiquitin-like modifier-activating enzyme 1 |
| Ube3a | Ubiquitin protein ligase E3A |
| UBL | Ubiquitin like protein |
| UPS | Ubiquitin-proteasome system |
| USE1 | UBA6 specific E2 |
| WISP1 | Wnt-induced secreted protein-1 |
| ZEB2 | Zinc finger E-box–binding homeobox 2 |
References
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Hershko, A.A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem. 2012, 81, 323–357. [Google Scholar] [CrossRef]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kalveram, B.; Raasi, S.; Groettrup, M.; Schmidtke, G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol. Cell Biol. 2005, 25, 3483–3491. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Sun, Q.; Chen, Z.J. E1-L2 activates both ubiquitin and FAT10. Mol. Cell 2007, 27, 1014–1023. [Google Scholar] [CrossRef]
- Aichem, A.; Pelzer, C.; Lukasiak, S.; Kalveram, B.; Sheppard, P.W.; Rani, N.; Schmidtke, G.; Groettrup, M. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat. Commun. 2010, 4, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Xi, J.; Begley, T.P.; Nicholson, L.K. Solution structure of ThiS and implications for the evolutionary roots of ubiquitin. Nat. Struct. Biol. 2001, 8, 47–51. [Google Scholar] [CrossRef]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Stoll, K.E.; Klevit, R.E. E2s: Structurally economical and functionally replete. Biochem. J. 2011, 433, 31–42. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014. [Google Scholar] [CrossRef]
- Groettrup, M.; Pelzer, C.; Schmidtke, G.; Hofmann, K. Activating the ubiquitin family: UBA6 challenges the field. Trends Biochem. Sci. 2008, 33, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, G.; Aichem, A.; Groettrup, M. FAT10ylation as a signal for proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol. Cell 2011, 43, 392–405. [Google Scholar] [CrossRef]
- Echchgadda, I.; Roth, C.C.; Cerna, C.Z.; Wilmink, G.J. Temporal gene expression kinetics for human keratinocytes exposed to hyperthermic stress. Cells 2013, 2, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Lange, N.; Urban, S.; Seifert, U.; Krüger, E.; Kloetzel, P.M. Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int. J. Biochem. Cell Biol. 2009, 41, 1205–1215. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kwak, M.; Lee, P.C. Impairment of social behavior and communication in mice lacking the Uba6-dependent ubiquitin activation system. Behav. Brain Res. 2015, 281, 78–85. [Google Scholar] [CrossRef]
- Lee, P.C.; Dodart, J.C.; Aron, L.; Finley, L.W.; Bronson, R.T.; Haigis, M.C.; Yankner, B.A.; Harper, J.W. Altered social behavior and neuronal development in mice lacking the Uba6-Use1 ubiquitin transfer system. Mol. Cell 2013, 50, 172–184. [Google Scholar] [CrossRef]
- Le Fevre, A.; Beygo, J.; Silveira, C.; Kamien, B.; Clayton-Smith, J.; Colley, A.; Buiting, K.; Dudding-Byth, T. Atypical Angelman syndrome due to a mosaic imprinting defect: Case reports and review of the literature. Am. J. Med. Genet. A 2017, 173, 753–757. [Google Scholar] [CrossRef]
- Louros, S.R.; Osterweil, E.K. Perturbed proteostasis in autism spectrum disorders. J. Neurochem. 2016, 139, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Cooper, E.M.; Hudson, A.W.; Amos, J.; Wagstaff, J.; Howley, P.M. Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem. 2004, 279, 41208–41217. [Google Scholar] [CrossRef] [PubMed]
- Day, C.; Shepherd, J.D. Arc: Building a bridge from viruses to memory. Biochem. J. 2015, 469, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, Y.; Kowa, H.; Tsuji, S.; Iwata, A. FAT10 protein binds to polyglutamine proteins and modulates their solubility. J. Biol. Chem. 2011, 286, 29594–29600. [Google Scholar] [CrossRef]
- French, S.W.; Mendoza, A.S.; Peng, Y. The mechanisms of Mallory-Denk body formation are similar to the formation of aggresomes in Alzheimer’s disease and other neurodegenerative disorders. Exp. Mol. Pathol. 2016, 100, 426–433. [Google Scholar] [CrossRef]
- Assawamakin, A.; Wattanasirichaigoon, D.; Tocharoentanaphol, C.; Waeteekul, S.; Tansatit, M.; Thongnoppakhun, W.; Limwongse, C. A novel maternally-derived insertional translocation resulting in partial trisomy 4q13.2-q22.1 with complex translocation t (8;20) in a family with intellectual disability. Am. J. Med. Genet. A 2012, 158A, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Matoso, E.; Melo, J.B.; Ferreira, S.I.; Jardim, A.; Castelo, T.M.; Weise, A.; Carreira, I.M. Insertional translocation leading to a 4q13 duplication including the EPHA5 gene in two siblings with attention-deficit hyperactivity disorder. Am. J. Med. Genet. A 2013, 161A, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Utine, G.E.; Haliloğlu, G.; Volkan-Salancı, B.; Çetinkaya, A.; Kiper, P.Ö.; Alanay, Y.; Aktaş, D.; Anlar, B.; Topçu, M.; Boduroğlu, K.; Alikaşifoğlu, M. Etiological yield of SNP microarrays in idiopathic intellectual disability. Eur. J. Paediatr. Neurol. 2014, 18, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Okamoto, N.; Nomura, S.; Fukui, M.; Shimakawa, S.; Sangu, N.; Shimojima, K.; Osawa, M.; Yamamoto, T. Microdeletions of 5.5 Mb (4q13.2-q13.3) and 4.1 Mb (7p15.3-p21.1) associated with a saethre-chotzen-like phenotype, severe intellectual disability, and autism. Am. J. Med. Genet. A 2013, 161A, 2078–2083. [Google Scholar] [CrossRef] [PubMed]
- Quintela, I.; Barros, F.; Fernandez-Prieto, M.; Martinez-Regueiro, R.; Castro-Gago, M.; Carracedo, A.; Gomez-Lado, C.; Eiris, J. Interstitial microdeletions including the chromosome band 4q13.2 and the UBA6 gene as possible causes of intellectual disability and behavior disorder. Am. J. Med. Genet. A 2015, 167A, 3113–3120. [Google Scholar] [CrossRef]
- Andreev, V.P.; Petyuk, V.A.; Brewer, H.M.; Karpievitch, Y.V.; Xie, F.; Clarke, J.; Camp, D.; Smith, R.D.; Lieberman, A.P.; Albin, R.L.; et al. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [Google Scholar] [CrossRef]
- Rubio, M.D.; Wood, K.; Haroutunian, V.; Meador-Woodruff, J.H. Dysfunction of the ubiquitin proteasome and ubiquitin-like systems in schizophrenia. Neuropsychopharmacology 2013, 38, 1910–1920. [Google Scholar] [CrossRef]
- Serpente, M.; Fenoglio, C.; Cioffi, S.M.; Bonsi, R.; Arighi, A.; Fumagalli, G.G.; Ghezzi, L.; Scarpini, E.; Galimberti, D. Profiling of ubiquitination pathway genes in peripheral cells from patients with frontotemporal dementia due to C9ORF72 and GRN mutations. Int. J. Mol. Sci. 2015, 16, 1385–1394. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Gursel, D.B.; Cheng, C.; Huang, S.; Rademaker, A.W.; Khan, S.A.; Yin, J.; Kiyokawa, H. The non-canonical ubiquitin activating enzyme UBA6 suppresses epithelial-mesenchymal transition of mammary epithelial cells. Oncotarget 2017, 8, 87480–87493. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, B.; Sun, L.; Bhuripanyo, K.; Wang, Y.; Bi, Y.; Davuluri, R.V.; Duong, D.M.; Nanavati, D.; Yin, J.; Kiyokawa, H. Orthogonal ubiquitin transfer identifies ubiquitination substrates under differential control by the two ubiquitin activating enzymes. Nat. Commun. 2017, 8, 14286. [Google Scholar] [CrossRef] [Green Version]
- Arpin, M.; Chirivino, D.; Naba, A.; Zwaenepoel, I. Emerging role for ERM proteins in cell adhesion and migration. Cell Adh. Migr. 2011, 5, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tepass, U. The apical polarity protein network in Drosophila epithelial cells: Regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu. Rev. Cell Dev. Biol. 2012, 28, 655–685. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodríguez-Pinilla, S.M.; Dotor, A.; Calero, F.; Hardisson, D.; Palacios, J. Abnormal ezrin localization is associated with clinicopathological features in invasive breast carcinomas. Breast Cancer Res. Treat. 2006, 98, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Buzzanco, A.; Gomez, A.; Rodriguez, E.; French, B.A.; Tillman, B.A.; Chang, S.; Ganapathy, E.; Junrungsee, S.; Zarrinpar, A.; Agopian, V.G.; et al. Digital quantitation of HCC-associated stem cell markers and protein quality control factors using tissue arrays of human liver sections. Exp. Mol. Pathol. 2014, 97, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.M.; Bowman, R.V.; Yang, I.A. Genomics of lung cancer. J. Thorac. Dis. 2017, 9, E155–E157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.R.; Rodriguez-Canales, J.; Gaffney, S.G.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2017, 28, 75–82. [Google Scholar] [CrossRef]
- Tonry, C.; Armstrong, J.; Pennington, S. Probing the prostate tumour microenvironment II: Impact of hypoxia on a cell model of prostate cancer progression. Oncotarget 2017, 8, 15307–15337. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Xu, Z.; Lu, W.; Li, X.; Sun, C.; Guo, J.; Xue, P.; Guan, F. Quantitative analysis of differential proteome expression in bladder cancer vs. normal bladder cells using SILAC method. PLoS ONE 2015, 10, e0134727. [Google Scholar] [CrossRef]
- Hogarth, C.A.; Mitchell, D.; Evanoff, R.; Smal, l.C.; Griswold, M. Identification and expression of potential regulators of the mammalian mitotic-to-meiotic transition. Biol. Reprod. 2011, 84, 34–42. [Google Scholar] [CrossRef]
- Anderson, E.L.; Baltus, A.E.; Roepers-Gajadien, H.L.; Hassold, T.J.; de Rooij, D.G.; van Pelt, A.M.; Page, D.C. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 14976–14980. [Google Scholar] [CrossRef] [Green Version]
- Schelpe, J.; Monté, D.; Dewitte, F.; Sixma, T.K.; Rucktooa, P. Structure of UBE2Z enzyme provides functional insight into specificity in the FAT10 protein conjugation machinery. J. Biol. Chem. 2016, 291, 630–639. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Zhao, B.; Bhuripanyo, K.; Zhang, K.; Kiyokawa, H.; Schindelin, H.; Yin, J. Orthogonal ubiquitin transfer through engineered E1-E2 cascades for protein ubiquitination. Chem. Biol. 2012, 19, 1265–1277. [Google Scholar] [CrossRef]
- Bates, E.E.; Ravel, O.; Dieu, M.C.; Ho, S.; Guret, C.; Bridon, J.M.; Ait-Yahia, S.; Brière, F.; Caux, C.; Banchereau, J.; Lebecque, S. Identification and analysis of a novel member of the ubiquitin family expressed in dendritic cells and mature B cells. Eur. J. Immunol. 1997, 27, 2471–2477. [Google Scholar] [CrossRef]
- Raasi, S.; Schmidtke, G.; de Giuli, R.; Groettrup, M. A ubiquitin-like protein which is synergistically inducible by interferon-gamma and tumor necrosis factor-alpha. Eur. J. Immunol. 1999, 29, 4030–4036. [Google Scholar] [CrossRef]
- Lim, C.B.; Zhang, D.; Lee, C.G. FAT10, a gene up-regulated in various cancers, is cell-cycle regulated. Cell Div. 2006, 1, 20. [Google Scholar] [CrossRef]
- Merbl, Y.; Refour, P.; Patel, H.; Springer, M.; Kirschner, M.W. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell 2013, 152, 1160–1172. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Buerger, S.; Groettrup, M. The ubiquitin-like modifier FAT10 in antigen processing and antimicrobial defense. Mol. Immunol. 2015, 68, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Cajee, U.F.; Hull, R.; Ntwasa, M. Modification by ubiquitin-like proteins: Significance in apoptosis and autophagy pathways. Int. J. Mol. Sci. 2012, 13, 11804–11831. [Google Scholar] [CrossRef]
- Aichem, A.; Groettrup, M. The ubiquitin-like modifier FAT10 in cancer development. Int. J. Biochem. Cell Biol. 2016, 79, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Yuan, R.; Wang, K.; Hu, J.; Yan, C.; Li, M.; Yu, X.; Liu, X.; Lei, J.; Guo, W.; Wu, L.; et al. Ubiquitin-like protein FAT10 promotes the invasion and metastasis of hepatocellular carcinoma by modifying β-catenin degradation. Cancer Res. 2014, 74, 5287–5300. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Ren, J.; Cheong, I.S.; Ban, K.H.; Ooi, L.L.; Yong, T.S.; Kan, A.; Nuchprayoon, I.; Jin, R.; Lee, K.H.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; French, B.A.; Oliva, J.; French, S.W. Increased expression of FAT10 in colon benign, premalignant and malignant epithelial neoplasms. Exp. Mol. Pathol. 2011, 90, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Theng, S.S.; Wang, W.; Mah, W.C.; Chan, C.; Zhuo, J.; Gao, Y.; Qin, H.; Lim, L.; Chong, S.S.; Song, J.; Lee, C.G. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2014, 111, E5282–E5291. [Google Scholar] [CrossRef]
- Canaan, A.; Yu, X.; Booth, C.J.; Lian, J.; Lazar, I.; Gamfi, S.L.; Castille, K.; Kohya, N.; Nakayama, Y.; Liu, Y.C.; et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol. Cell Biol. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Canaan, A.; DeFuria, J.; Perelman, E.; Schultz, V.; Seay, M.; Tuck, D.; Flavell, R.A.; Snyder, M.P.; Obin, M.S.; Weissman, S.M. Extended lifespan and reduced adiposity in mice lacking the FAT10 gene. Proc. Natl. Acad. Sci. USA 2014, 111, 5313–5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Cai, W.; Parimoo, S.; Schwarz, D.C.; Lennon, G.G.; Weissman, S.M. Identification of seven new human MHC class I region genes around the HLA-F locus. Immunogenetics 1996, 44, 97–103. [Google Scholar] [CrossRef]
- Aichem, A.; Anders, S.; Catone, N.; Rößler, P.; Stotz, S.; Berg, A.; Schwab, R.; Scheuermann, S.; Bialas, J.; Schütz-Stoffregen, M.C.; et al. The structure of the ubiquitin-like modifier FAT10 reveals an alternative targeting mechanism for proteasomal degradation. Nat. Commun. 2018, 9, 3321. [Google Scholar] [CrossRef]
- Rani, N.; Aichem, A.; Schmidtke, G.; Kreft, S.G.; Groettrup, M. FAT10 and NUB1L bind to the VWA domain of Rpn10 and Rpn1 to enable proteasome-mediated proteolysis. Nat. Commun. 2012, 3, 749. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, G.; Kalveram, B.; Weber, E.; Bochtler, P.; Lukasiak, S.; Hipp, M.S.; Groettrup, M. The UBA domains of NUB1L are required for binding but not for accelerated degradation of the ubiquitin-like modifier FAT10. J. Biol. Chem. 2006, 281, 20045–20054. [Google Scholar] [CrossRef]
- Aichem, A.; Catone, N.; Groettrup, M. Investigations into the auto-FAT10ylation of the bispecific E2 conjugating enzyme UBA6-specific E2 enzyme 1. FEBS J. 2014, 281, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Raasi, S.; Groettrup, M.; Schmidtke, G. NEDD8 ultimate buster-1L interacts with the ubiquitin-like protein FAT10 and accelerates its degradation. J. Biol. Chem. 2004, 279, 16503–16510. [Google Scholar] [CrossRef]
- Schmidtke, G.; Kalveram, B.; Groettrup, M. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett. 2009, 583, 591–594. [Google Scholar] [CrossRef] [Green Version]
- Bett, J.S.; Kanuga, N.; Richet, E.; Schmidtke, G.; Groettrup, M.; Cheetham, M.E.; van der Spuy, J. The inherited blindness protein AIPL1 regulates the ubiquitin-like FAT10 pathway. PLoS ONE 2012, 7, e30866. [Google Scholar] [CrossRef]
- Akey, D.T.; Zhu, X.; Dyer, M.; Li, A.; Sorensen, A.; Blackshaw, S.; Fukuda-Kamitani, T.; Daiger, S.P.; Craft, C.M.; Kamitani, T.; Sohocki, M.M. The inherited blindness associated protein AIPL1 interacts with the cell cycle regulator protein NUB1. Hum. Mol. Genet. 2002, 11, 2723–2733. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Liu, Y.; Gu, X.; Zhu, T.; Yang, S.; Sun, W. LMO2 blocks the UBA6-USE1 interaction and downstream FAT10ylation by targeting the ubiquitin fold domain of UBA6. Biochem. Biophys. Res. Commun. 2016, 478, 1442–1448. [Google Scholar] [CrossRef]
- Aichem, A.; Kalveram, B.; Spinnenhirn, V.; Kluge, K.; Catone, N.; Johansen, T.; Groettrup, M. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J. Cell Sci. 2012, 125, 4576–4585. [Google Scholar] [CrossRef] [Green Version]
- Leng, L.; Xu, C.; Wei, C.; Zhang, J.; Liu, B.; Ma, J.; Li, N.; Qin, W.; Zhang, W.; Zhang, C.; et al. A proteomics strategy for the identification of FAT10-modified sites by mass spectrometry. J. Proteome Res. 2014, 13, 268–276. [Google Scholar] [CrossRef]
- Bialas, J.; Boehm, A.N.; Catone, N.; Aichem, A.; Groettrup, M. The ubiquitin-like modifier FAT10 stimulates the activity of the deubiquitylating enzyme OTUB1. J. Biol. Chem. 2019, 294, 4315–4330. [Google Scholar] [CrossRef]
- Yan, J.; Lei, J.; Chen, L.; Deng, H.; Dong, D.; Jin, T.; Liu, X.; Yuan, R.; Qiu, Y.; Ge, J.; et al. Human leukocyte antigen F locus adjacent transcript 10 overexpression disturbs WISP1 protein and mRNA expression to promote hepatocellular carcinoma progression. Hepatology 2018, 68, 2268–2284. [Google Scholar] [CrossRef]
- Li, T.; Santockyte, R.; Yu, S.; Shen, R.F.; Tekle, E.; Lee, C.G.; Yang, D.C.; Chock, P.B. FAT10 modifies p53 and upregulates its transcriptional activity. Arch. Biochem. Biophys. 2011, 509, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Kim, J.K.; Yoo, J.Y. NFκB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Mol. Oncol. 2014, 8, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.W.; Jeang, K.T.; Lee, C.G. p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene 2006, 25, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Bialas, J.; Groettrup, M.; Aichem, A. Conjugation of the ubiquitin activating enzyme UBE1 with the ubiquitin-like modifier FAT10 targets it for proteasomal degradation. PLoS ONE 2015, 10, e0120329. [Google Scholar] [CrossRef]
- Kalveram, B.; Schmidtke, G.; Groettrup, M. The ubiquitin-like modifier FAT10 interacts with HDAC6 and localizes to aggresomes under proteasome inhibition. J. Cell Sci. 2008, 121, 4079–4088. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- French, S.W.; French, B.A.; Oliva, J.; Li, J.; Bardag-Gorce, F.; Tillman, B.; Canaan, A. FAT10 knock out mice livers fail to develop Mallory-Denk bodies in the DDC mouse model. Exp. Mol. Pathol. 2012, 93, 309–314. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Oliva, J.; Li, J.; French, B.A.; French, S.W. SAMe prevents the induction of the immunoproteasome and preserves the 26S proteasome in the DDC-induced MDB mouse model. Exp. Mol. Pathol. 2010, 88, 353–362. [Google Scholar] [CrossRef]
- Oliva, J.; Bardag-Gorce, F.; Li, J.; French, B.A.; Nguyen, S.K.; Lu, S.C.; French, S.W. Betaine prevents Mallory-Denk body formation in drug-primed mice by epigenetic mechanisms. Exp. Mol. Pathol. 2009, 86, 77–86. [Google Scholar] [CrossRef]
- French, S.W.; Masouminia, M.; Samadzadeh, S.; Tillman, B.C.; Mendoza, A.; French, B.A. Role of protein quality control failure in alcoholic hepatitis pathogenesis. Biomolecules 2017, 7, 11. [Google Scholar] [CrossRef]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef]
- Hiroi, Y.; Rechsteiner, M. Ubiquitin metabolism in HeLa cells starved of amino acids. FEBS Lett. 1992, 307, 156–161. [Google Scholar] [CrossRef] [Green Version]
- van den Boom, J.; Meyer, H. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Gavin, J.M.; Chen, J.J.; Liao, H.; Rollins, N.; Yang, X.; Xu, Q.; Ma, J.; Loke, H.K.; Lingaraj, T.; Brownell, J.E.; et al. Mechanistic studies on activation of ubiquitin and di-ubiquitin-like protein, FAT10, by ubiquitin-like modifier activating enzyme 6, Uba6. J. Biol. Chem. 2012, 287, 15512–15522. [Google Scholar] [CrossRef]
- Buchsbaum, S.; Bercovich, B.; Ciechanover, A. FAT10 is a proteasomal degradation signal that is itself regulated by ubiquitination. Mol. Biol. Cell 2012, 23, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Wiener, R.; DiBello, A.T.; Lombardi, P.M.; Guzzo, C.M.; Zhang, X.; Matunis, M.J.; Wolberger, C. E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat. Struct. Mol. Biol. 2013, 20, 1033–1039. [Google Scholar] [CrossRef]
- Zhou, Q.; Peng, X.; Liu, X.; Chen, L.; Xiong, Q.; Shen, Y.; Xie, J.; Xu, Z.; Huang, L.; Hu, J.; et al. FAT10 attenuates hypoxia-induced cardiomyocyte apoptosis by stabilizing caveolin-3. J. Mol. Cell Cardiol. 2018, 116, 115–124. [Google Scholar] [CrossRef]
- Zou, Y.; Ouyang, Q.; Wei, W.; Yang, S.; Zhang, Y.; Yang, W. FAT10 promotes the invasion and migration of breast cancer cell through stabilization of ZEB2. Biochem. Biophys. Res. Commun. 2018, 506, 563–570. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Ge, J.; Yan, C.; Huang, Z.; Hu, J.; Wen, C.; Li, M.; Huang, D.; Qiu, Y.; et al. The ubiquitin-like protein FAT10 stabilizes eEF1A1 expression to promote tumor proliferation in a complex manner. Cancer Res. 2016, 76, 4897–4907. [Google Scholar] [CrossRef]
- Gu, X.; Zhao, F.; Zheng, M.; Fei, X.; Chen, X.; Huang, S.; Xie, Y.; Mao, Y. Cloning and characterization of a gene encoding the human putative ubiquitin conjugating enzyme E2Z (UBE2Z). Mol. Biol. Rep. 2007, 34, 183–188. [Google Scholar] [CrossRef]
- van Wijk, S.J.; Timmers, H.T. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, F.; Li, L.; Gao, H.; Arckacki, S.; Wang, I.Z.; Barnard, J.; Ellis, S.; Hubbard, C.; Topol, E.J.; et al. Genome-wide linkage analysis of large multiple multigenerational families identifies novel genetic loci for coronary artery disease. Sci. Rep. 2017, 7, 5472. [Google Scholar] [CrossRef]
- Kessler, T.; Vilne, B.; Schunkert, H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol. Med. 2016, 8, 688–701. [Google Scholar] [CrossRef]
- Han, Y.; Dorajoo, R.; Chang, X.; Wang, L.; Khor, C.C.; Sim, X.; Cheng, C.Y.; Shi, Y.; Tham, Y.C.; Zhao, W.; et al. Genome-wide association study identifies a missense variant at APOA5 for coronary artery disease in Multi-Ethnic Cohorts from Southeast Asia. Sci. Rep. 2017, 7, 17921. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Huang, J.; Ma, X.; Gu, N.; Zhang, J.; Zhang, H.; Guo, X. Rs46522 in the ubiquitin-conjugating enzyme E2Z gene is associated with the risk of coronary artery disease in individuals of Chinese Han population with type 2 diabetes. J. Diabetes Res. 2017. [Google Scholar] [CrossRef]
- Bastami, M.; Ghaderian, S.M.; Omrani, M.D.; Mirfakhraie, R.; Nariman-Saleh-Fam, Z.; Mansoori, Y.; Masotti, A. Evaluating the association of common UBE2Z variants with coronary artery disease in an Iranian population. Cell Mol. Biol (Noisy-le-grand) 2015, 61, 50–54. [Google Scholar]
- Hartmann, K.; Seweryn, M.; Handelman, S.K.; Rempała, G.A.; Sadee, W. Non-linear interactions between candidate genes of myocardial infarction revealed in mRNA expression profiles. BMC Genom. 2016, 17, 738. [Google Scholar] [CrossRef]
- Brænne, I.; Civelek, M.; Vilne, B.; Di Narzo, A.; Johnson, A.D.; Zhao, Y.; Reiz, B.; Codoni, V.; Webb, T.R.; Foroughi Asl, H. Prediction of causal candidate genes in coronary artery disease Loci. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2207–2217. [Google Scholar] [CrossRef]
- Dehghan, A.; Bis, J.C.; White, C.C.; Smith, A.V.; Morrison, A.C.; Cupples, L.A.; Trompet, S.; Chasman, D.I.; Lumley, T.; Völker, U.; et al. Genome-wide association study for incident myocardial infarction and coronary heart disease in prospective cohort studies: The CHARGE consortium. PLoS ONE 2016, 11, e0144997. [Google Scholar] [CrossRef]
- Zhang, H.; Wheeler, W.; Hyland, P.L.; Yang, Y.; Shi, J.; Chatterjee, N.; Yu, K. A powerful procedure for pathway-based meta-analysis using summary statistics identifies 43 pathways associated with type II diabetes in European populations. PLoS Genet. 2016, 12, e1006122. [Google Scholar] [CrossRef]
- Bonàs-Guarch, S.; Guindo-Martínez, M.; Miguel-Escalada, I.; Grarup, N.; Sebastian, D.; Rodriguez-Fos, E.; Sánchez, F.; Planas-Fèlix, M.; Cortes-Sánchez, P.; González, S.; et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat. Commun. 2018, 9, 321. [Google Scholar] [CrossRef]
- Kim, S.J.; Hyeong Lee, T.; Hee Nam, S.; Kim, J.H.; Oh, S.; Sook Cho, Y.; Sup Lee, M.; Choi, S.; Lee, P.C. Association of Uba6-Specific-E2 (USE1) with lung tumorigenesis. J. Natl. Cancer Inst. 2017, 109, 1–11. [Google Scholar] [CrossRef]
- Xu, W.; Neckers, L. A USE1ful biomarker and molecular target in Lung Cancer? J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Huang, G.; Song, H.; Wang, R.; Han, X.; Chen, L. The relationship between RGS5 expression and cancer differentiation and metastasis in non-small cell lung cancer. J. Surg. Oncol. 2012, 105, 420–424. [Google Scholar] [CrossRef]
- Cheng, C.; Yue, W.; Li, L.; Li, S.; Gao, C.; Si, L.; Tian, H. Regulator of G-protein signaling 4: A novel tumor suppressor with prognostic significance in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2016, 469, 384–391. [Google Scholar] [CrossRef]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zhao, B. UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10. Int. J. Mol. Sci. 2019, 20, 2250. https://doi.org/10.3390/ijms20092250
Wang F, Zhao B. UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10. International Journal of Molecular Sciences. 2019; 20(9):2250. https://doi.org/10.3390/ijms20092250
Chicago/Turabian StyleWang, Fengting, and Bo Zhao. 2019. "UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10" International Journal of Molecular Sciences 20, no. 9: 2250. https://doi.org/10.3390/ijms20092250
APA StyleWang, F., & Zhao, B. (2019). UBA6 and Its Bispecific Pathways for Ubiquitin and FAT10. International Journal of Molecular Sciences, 20(9), 2250. https://doi.org/10.3390/ijms20092250