Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
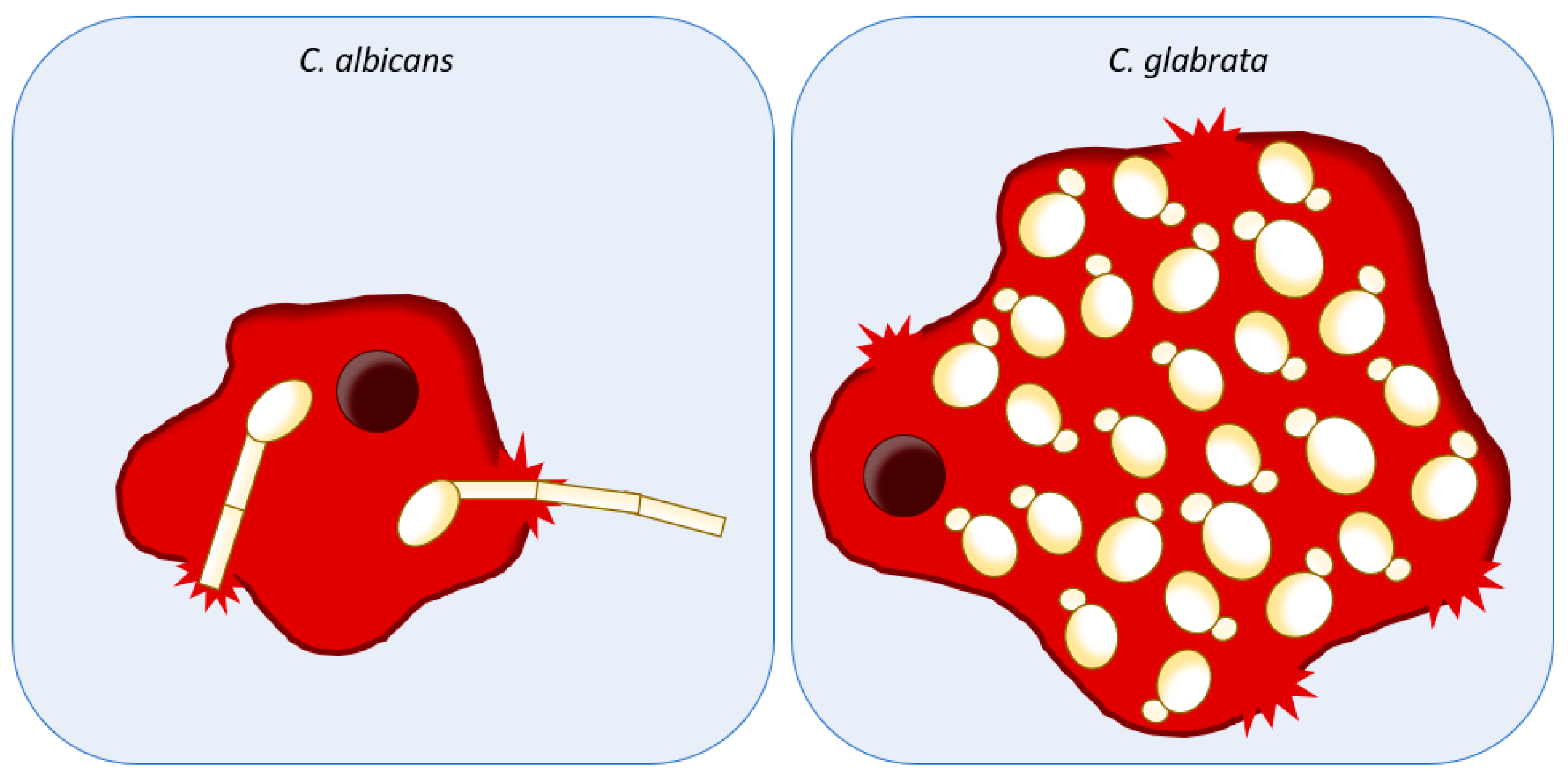
2. Host Damage and Invasion
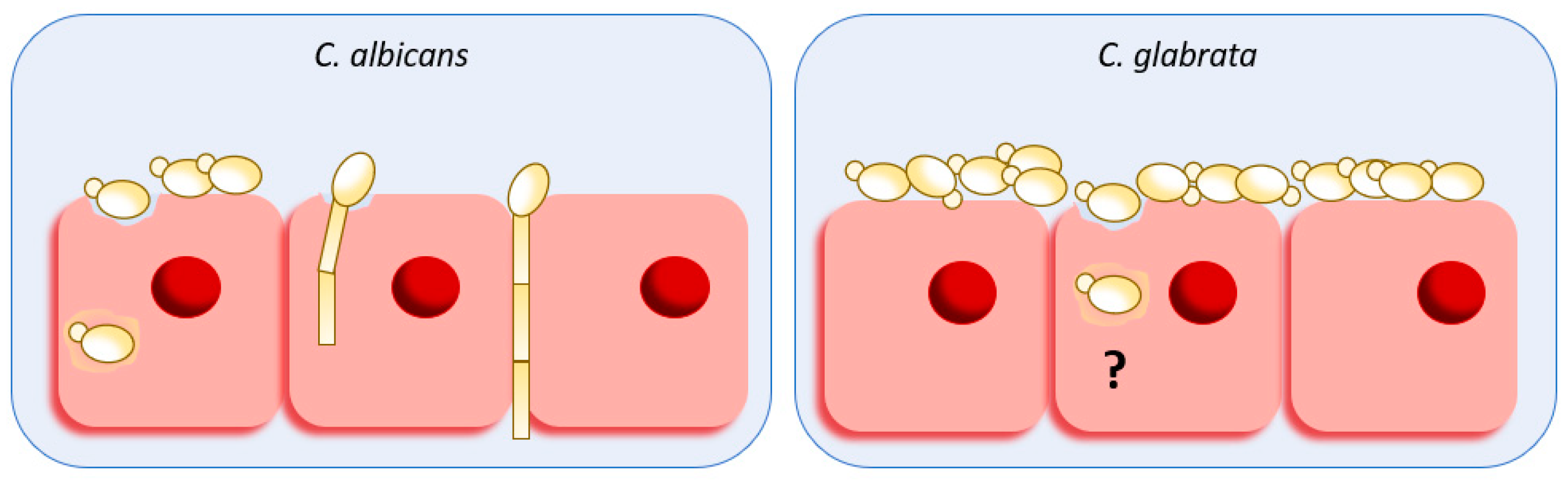
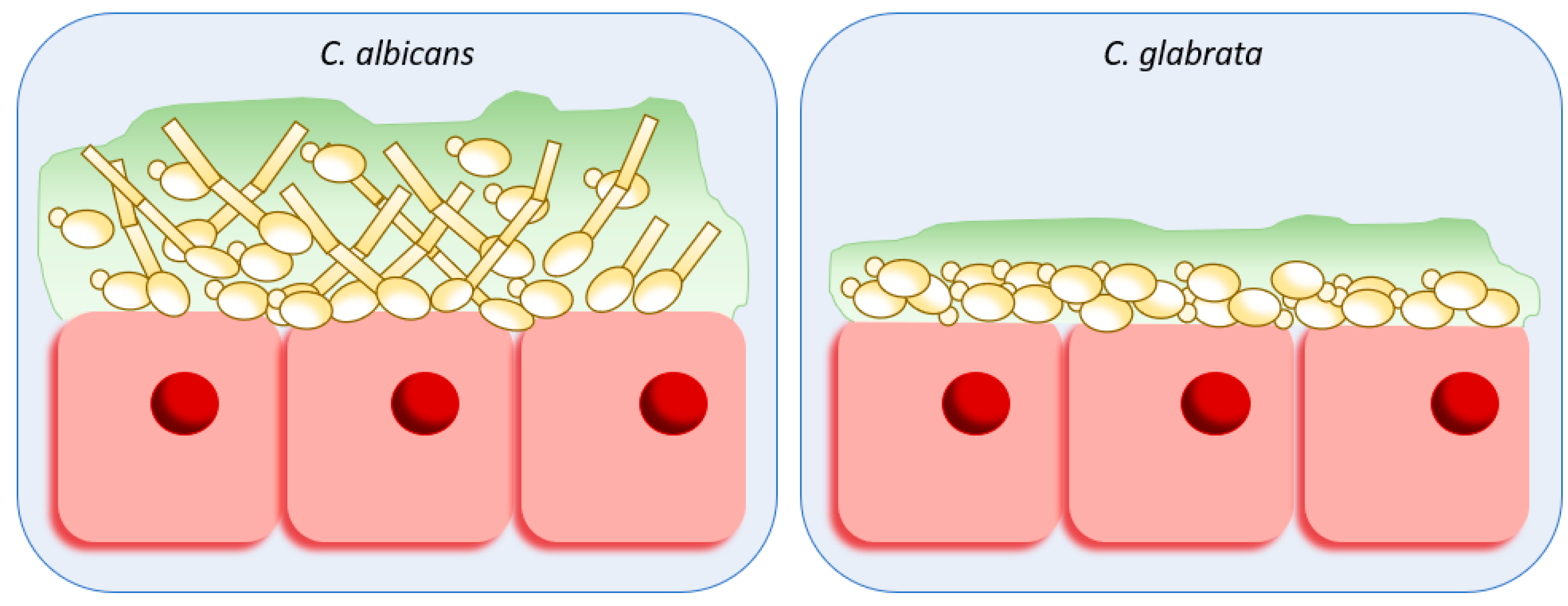
3. Adhesion and Biofilm Formation
4. Host Immune System Evasion
5. Conclusion and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Peghin, M.; Timsit, J.-F. The current treatment landscape: Candidiasis. J. Antimicrob. Chemother. 2016, 71, ii13–ii22. [Google Scholar] [CrossRef]
- Olson, M.L.; Jayaraman, A.; Kao, K.C. Relative Abundances of Candida albicans and Candida glabrata in In Vitro Coculture Biofilms Impact Biofilm Structure and Formation. Appl. Environ. Microbiol. 2018, 84, e02769-17. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial Bloodstream Infections in US Hospitals: Analysis of 24,179 Cases from a Prospective Nationwide Surveillance Study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.; Federspiel, N.A.; Chibana, H.; Dungan, J.; Kalman, S.; Magee, B.B.; Newport, G.; Thorstenson, Y.R.; Agabian, N.; Magee, P.T.; et al. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. USA 2004, 101, 7329–7334. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L. The ALS gene family of Candida albicans. Trends Microbiol. 2001, 9, 176–180. [Google Scholar] [CrossRef]
- Ghannoum, M.A. Potential role of phospholipases in virulence and fungal pathogenesis. Clin. Microbiol. Rev. 2000, 13, 122–143. [Google Scholar] [CrossRef] [PubMed]
- Calderone, R.A.; Fonzi, W.A. Virulence factors of Candida albicans. Trends Microbiol. 2001, 9, 327–335. [Google Scholar] [CrossRef]
- Li, L.; Kashleva, H.; Dongari-Bagtzoglou, A. Cytotoxic and cytokine-inducing properties of Candida glabrata in single and mixed oral infection models. Microb. Pathog. 2007, 42, 138–147. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.W.J.; Kraneveld, E.A.; Yin, Q.Y.; Dekker, H.L.; Groß, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The Cell Wall of the Human Pathogen Candida glabrata: Differential Incorporation of Novel Adhesin-Like Wall Proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar] [CrossRef]
- Kaur, R.; Ma, B.; Cormack, B.P. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. USA 2007, 104, 7628–7633. [Google Scholar] [CrossRef]
- Kaur, R.; Domergue, R.; Zupancic, M.L.; Cormack, B.P. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 2005, 8, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Brunke, S.; Hube, B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell. Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.; Seider, K.; Hube, B. Intracellular survival of Candida glabrata in macrophages: Immune evasion and persistence. FEMS Yeast Res. 2015, 15, fov042. [Google Scholar] [CrossRef] [PubMed]
- Uwamahoro, N.; Verma-Gaur, J.; Shen, H.H.; Qu, Y.; Lewis, R.; Lu, J.; Bambery, K.; Masters, S.L.; Vince, J.E.; Naderer, T.; et al. The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. MBio 2014, 5, e00003-14. [Google Scholar] [CrossRef]
- Lorenz, M.C.; Bender, J.A.; Fink, G.R. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 2004, 3, 1076–1087. [Google Scholar] [CrossRef]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef]
- Cavalheiro, M.; Teixeira, M.C. Candida Biofilms: Threats, Challenges, and Promising Strategies. Front. Med. 2018, 5, 28. [Google Scholar] [CrossRef]
- Seider, K.; Brunke, S.; Schild, L.; Jablonowski, N.; Wilson, D.; Majer, O.; Barz, D.; Haas, A.; Kuchler, K.; Schaller, M.; et al. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J. Immunol. 2011, 187, 3072–3086. [Google Scholar] [CrossRef]
- Dementhon, K.; El-Kirat-Chatel, S.; Noël, T. Development of an in vitro model for the Multi-Parametric quantification of the cellular interactions between Candida yeasts and phagocytes. PLoS ONE 2012, 7, e32621. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Gratz, N.; Kovarik, P.; Schãller, C. Autophagy supports Candida glabrata survival during phagocytosis. Cell. Microbiol. 2010, 12, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yang, C.; Tang, J. Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol. Res. 2013, 168, 389–395. [Google Scholar] [CrossRef]
- Allert, S.; Förster, T.M.; Svensson, C.-M.; Richardson, J.P.; Pawlik, T.; Hebecker, B.; Rudolphi, S.; Juraschitz, M.; Schaller, M.; Blagojevic, M.; et al. Candida albicans-Induced Epithelial Damage Mediates Translocation through Intestinal Barriers. MBio 2018, 9, e00915-18. [Google Scholar] [CrossRef]
- Cassone, A. Vulvovaginal Candida albicans infections: Pathogenesis, immunity and vaccine prospects. BJOG Int. J. Obstet. Gynaecol. 2015, 122, 785–794. [Google Scholar] [CrossRef]
- Bokor-Bratic, M.; Cankovic, M.; Dragnic, N. Unstimulated whole salivary flow rate and anxiolytics intake are independently associated with oral Candida infection in patients with oral lichen planus. Eur. J. Oral Sci. 2013, 121, 427–433. [Google Scholar] [CrossRef]
- Hibino, K.; Samaranayake, L.P.; Hägg, U.; Wong, R.W.K.; Lee, W. The role of salivary factors in persistent oral carriage of Candida in humans. Arch. Oral Biol. 2009, 54, 678–683. [Google Scholar] [CrossRef]
- Goyer, M.; Loiselet, A.; Bon, F.; L’Ollivier, C.; Laue, M.; Holland, G.; Bonnin, A.; Dalle, F. Intestinal Cell Tight Junctions Limit Invasion of Candida albicans through Active Penetration and Endocytosis in the Early Stages of the Interaction of the Fungus with the Intestinal Barrier. PLoS ONE 2016, 11, e0149159. [Google Scholar] [CrossRef]
- Dalle, F.; Wãchtler, B.; L’Ollivier, C.; Holland, G.; Bannert, N.; Wilson, D.; Labruãre, C.; Bonnin, A.; Hube, B. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell. Microbiol. 2010, 12, 248–271. [Google Scholar] [CrossRef]
- Phan, Q.T.; Myers, C.L.; Fu, Y.; Sheppard, D.C.; Yeaman, M.R.; Welch, W.H.; Ibrahim, A.S.; Edwards, J.E.; Filler, S.G. Als3 Is a Candida albicans Invasin That Binds to Cadherins and Induces Endocytosis by Host Cells. PLoS Biol. 2007, 5, e64. [Google Scholar] [CrossRef]
- Sun, J.N.; Solis, N.V.; Phan, Q.T.; Bajwa, J.S.; Kashleva, H.; Thompson, A.; Liu, Y.; Dongari-Bagtzoglou, A.; Edgerton, M.; Filler, S.G. Host Cell Invasion and Virulence Mediated by Candida albicans Ssa1. PLoS Pathog. 2010, 6, e1001181. [Google Scholar] [CrossRef]
- Zakikhany, K.; Naglik, J.R.; Schmidt-Westhausen, A.; Holland, G.; Schaller, M.; Hube, B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell. Microbiol. 2007, 9, 2938–2954. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Myers, C.L.; Sheppard, D.C.; Phan, Q.T.; Sanchez, A.A.; Edwards, J.E.; Filler, S.G. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell. Microbiol. 2004, 7, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Nielsen, K. Morphology Changes in Human Fungal Pathogens upon Interaction with the Host. J. Fungi 2017, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, I.D.; Wilson, D.; Wächtler, B.; Brunke, S.; Naglik, J.R.; Hube, B. Candida albicans dimorphism as a therapeutic target. Expert Rev. Anti-Infect. Ther. 2012, 10, 85–93. [Google Scholar] [CrossRef]
- Feng, Q.; Summers, E.; Guo, B.; Fink, G. Ras signaling is required for serum-induced hyphal differentiation in Candida albicans. J. Bacteriol. 1999, 181, 6339–6346. [Google Scholar]
- Maidan, M.M.; De Rop, L.; Serneels, J.; Exler, S.; Rupp, S.; Tournu, H.; Thevelein, J.M.; Van Dijck, P. The G Protein-coupled Receptor Gpr1 and the Gα Protein Gpa2 Act through the cAMP-Protein Kinase A Pathway to Induce Morphogenesis in Candida albicans. Mol. Biol. Cell 2005, 16, 1971–1986. [Google Scholar] [CrossRef]
- Biswas, K.; Morschhäuser, J. The Mep2p ammonium permease controls nitrogen starvation-induced filamentous growth in Candida albicans. Mol. Microbiol. 2005, 56, 649–669. [Google Scholar] [CrossRef]
- Stoldt, V.R.; Sonneborn, A.; Leuker, C.E.; Ernst, J.F. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 1997, 16, 1982–1991. [Google Scholar] [CrossRef]
- Zeidler, U.; Lettner, T.; Lassnig, C.; Müller, M.; Lajko, R.; Hintner, H.; Breitenbach, M.; Bito, A. UME6 is a crucial downstream target of other transcriptional regulators of true hyphal development in Candida albicans. FEMS Yeast Res. 2009, 9, 126–142. [Google Scholar] [CrossRef]
- Murad, A.M.A.; Leng, P.; Straffon, M.; Wishart, J.; Macaskill, S.; MacCallum, D.; Schnell, N.; Talibi, D.; Marechal, D.; Tekaia, F.; et al. NRG1 represses yeast-hypha morphogenesis and hypha-specific gene expression in Candida albicans. EMBO J. 2001, 20, 4742–4752. [Google Scholar] [CrossRef]
- Braun, B.R.; Kadosh, D.; Johnson, A.D. NRG1, a repressor of filamentous growth in C.albicans, is down-regulated during filament induction. EMBO J. 2001, 20, 4753–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, H.J.; Veacutery, A.-A.; Perera, T.H.S.; Davies, J.M.; Gow, N.A.R. Thigmotropism and stretch-activated channels in the pathogenic fungus Candida albicans. Microbiology 1998, 144, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Tscherner, M.; Giessen, T.W.; Markey, L.; Kumamoto, C.A.; Silver, P.A. A Synthetic System That Senses Candida albicans and Inhibits Virulence Factors. ACS Synth. Biol. 2019, 8, 434–444. [Google Scholar] [CrossRef]
- Chen, H.; Fujita, M.; Feng, Q.; Clardy, J.; Fink, G.R. Tyrosol is a quorum-sensing molecule in Candida albicans. Proc. Natl. Acad. Sci. USA 2004, 101, 5048–5052. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.R.; Rodgers, C.A.; Shirlaw, P.J.; Dobbie, J.L.; Fernandes-Naglik, L.L.; Greenspan, D.; Agabian, N.; Challacombe, S.J. Differential Expression of Candida albicans Secreted Aspartyl Proteinase and Phospholipase B Genes in Humans Correlates with Active Oral and Vaginal Infections. J. Infect. Dis. 2003, 188, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Hube, B.; Naglik, J. Candida albicans proteinases: Resolving the mystery of a gene family. Microbiology 2001, 147, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Borg-von Zepelin, M.; Beggah, S.; Boggian, K.; Sanglard, D.; Monod, M. The expression of the secreted aspartyl proteinases Sap4 to Sap6 from Candida albicans in murine macrophages. Mol. Microbiol. 1998, 28, 543–554. [Google Scholar] [CrossRef]
- Bizzarri, M.; Fuso, A.; Dinicola, S.; Cucina, A.; Bevilacqua, A. Pharmacodynamics and pharmacokinetics of inositol(s) in health and disease. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1181–1196. [Google Scholar] [CrossRef]
- Tsang, P.W.-K.; Fong, W.-P.; Samaranayake, L.P. Candida albicans orf19.3727 encodes phytase activity and is essential for human tissue damage. PLoS ONE 2017, 12, e0189219. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Lim, J.K.; Lee, C.-C.R.; Murphy, P.M. Organ-Specific Innate Immune Responses in a Mouse Model of Invasive Candidiasis. J. Innate Immunity 2011, 3, 180–199. [Google Scholar] [CrossRef]
- Saville, S.P.; Lazzell, A.L.; Monteagudo, C.; Lopez-Ribot, J.L. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot. Cell 2003, 2, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Csank, C.; Haynes, K. Candida glabrata displays pseudohyphal growth. FEMS Microbiol. Lett. 2000, 189, 115–120. [Google Scholar] [CrossRef]
- Sasani, E.; Khodavaisy, S.; Agha Kuchak Afshari, S.; Darabian, S.; Aala, F.; Rezaie, S. Pseudohyphae formation in Candida glabrata due to CO2 exposure. Curr. Med. Mycol. 2016, 2, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Perlroth, J.; Choi, B.; Spellberg, B. Nosocomial fungal infections: Epidemiology, diagnosis, and treatment. Med. Mycol. 2007, 45, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, R.; Angoulvant, A.; Tefit, M.; Gay, F.; Guitard, J.; Mazier, D.; Fairhead, C.; Hennequin, C. A mouse model for Candida glabrata hematogenous disseminated infection starting from the gut: Evaluation of strains with different adhesion properties. PLoS ONE 2013, 8, e69664. [Google Scholar] [CrossRef]
- Jacobsen, I.D.; Große, K.; Berndt, A.; Hube, B. Pathogenesis of Candida albicans infections in the alternative chorio-allantoic membrane chicken embryo model resembles systemic murine infections. PLoS ONE 2011, 6, e19741. [Google Scholar] [CrossRef]
- Silva, S.; Henriques, M.C.; Hayes, A.; Oliveira, R.; Azeredo, J.; Williams, D.W. Candida glabrata and Candida albicans co-infection of an in vitro oral epithelium. J. Oral Pathol. Med. 2011, 40, 421–427. [Google Scholar] [CrossRef]
- Coco, B.J.; Bagg, J.; Cross, L.J.; Jose, A.; Cross, J.; Ramage, G. Mixed Candida albicans and Candida glabrata populations associated with the pathogenesis of denture stomatitis. Oral Microbiol. Immunol. 2008, 23, 377–383. [Google Scholar] [CrossRef]
- Okada, K.; Nakazawa, S.; Yokoyama, A.; Kashiwazaki, H.; Kobayashi, K.; Yamazaki, Y. A Clinical Study of Candida albicans and Candida glabrata Co-infection of Oral Candidiasis. Ronen Shika Igaku 2016, 31, 346–353. [Google Scholar]
- Tsay, S.; Williams, S.R.; Benedict, K.; Beldavs, Z.; Farley, M.; Harrison, L.; Schaffner, W.; Dumyati, G.; Blackstock, A.; Guh, A.; et al. A Tale of Two Healthcare-associated Infections: Clostridium difficile Coinfection Among Patients With Candidemia. Clin. Infect. Dis. 2018, 64, 676–679. [Google Scholar] [CrossRef]
- Tati, S.; Davidow, P.; McCall, A.; Hwang-Wong, E.; Rojas, I.G.; Cormack, B.; Edgerton, M. Candida glabrata Binding to Candida albicans Hyphae Enables Its Development in Oropharyngeal Candidiasis. PLoS Pathog. 2016, 12, e1005522. [Google Scholar] [CrossRef]
- Alves, C.T.; Wei, X.Q.; Silva, S.; Azeredo, J.; Henriques, M.; Williams, D.W. Candida albicans promotes invasion and colonisation of Candida glabrata in a reconstituted human vaginal epithelium. J. Infect. 2014, 69, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dongari-Bagtzoglou, A. Epithelial GM-CSF induction by Candida glabrata. J. Dent. Res. 2009, 88, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L.; Vazquez, J.A.; Sobel, J.D. Candida glabrata: Review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 1999, 12, 80–96. [Google Scholar] [CrossRef]
- Fatahinia, M.; Halvaeezadeh, M.; Rezaei-Matehkolaei, A. Comparison of enzymatic activities in different Candida species isolated from women with vulvovaginitis. J. Mycol. Med. 2017, 27, 188–194. [Google Scholar] [CrossRef]
- Bassyouni, R.H.; Wegdan, A.A.; Abdelmoneim, A.; Said, W.; Aboelnaga, F. Phospholipase and aspartyl proteinase activities of candida species causing vulvovaginal candidiasis in patients with type 2 diabetes mellitus. J. Microbiol. Biotechnol. 2015, 25, 1734–1741. [Google Scholar] [CrossRef]
- De Riceto, É.B.M.; de Menezes, R.P.; Penatti, M.P.A.; dos Pedroso, R.S. Enzymatic and hemolytic activity in different Candida species. Rev. Iberoam. Micol. 2015, 32, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Atalay, M.A.; Koc, A.N.; Demir, G.; Sav, H. Investigation of possible virulence factors in Candida strains isolated from blood cultures. Niger. J. Clin. Pract. 2015, 18, 52–55. [Google Scholar] [PubMed]
- Pandey, N.; Gupta, M.K.; Tilak, R. Extracellular hydrolytic enzyme activities of the different Candida spp. isolated from the blood of the Intensive Care Unit-admitted patients. J. Lab. Phys. 2018, 10, 392–396. [Google Scholar]
- Canela, H.M.S.; Cardoso, B.; Vitali, L.H.; Coelho, H.C.; Martinez, R.; da Ferreira, M.E.S. Prevalence, virulence factors and antifungal susceptibility of Candida spp. isolated from bloodstream infections in a tertiary care hospital in Brazil. Mycoses 2018, 61, 11–21. [Google Scholar] [CrossRef]
- Rossoni, R.D.; Barbosa, J.O.; Vilela, S.F.G.; Dos Santos, J.D.; Jorge, A.O.C.; Junqueira, J.C. Correlation of phospholipase and proteinase production of Candida with in vivo pathogenicity in Galleria mellonella. Braz. J. Oral Sci. 2013, 12, 199–204. [Google Scholar] [CrossRef]
- Naglik, J.R.; Challacombe, S.J.; Hube, B. Candida albicans Secreted Aspartyl Proteinases in Virulence and Pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 400–428. [Google Scholar] [CrossRef]
- Parra-Ortega, B.; Cruz-Torres, H.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Phylogeny and evolution of the aspartyl protease family from clinically relevant Candida species. Mem. Inst. Oswaldo Cruz 2009, 104, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.F.; Silva, S.; Henriques, M. Candida glabrata: A review of its features and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Chumber, S.; Kaur, M. Studying the prevalence, species distribution, and detection of in vitro production of phospholipase from Candida isolated from cases of invasive Candidiasis. J. Glob. Infect. Dis. 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Rodrigues, M.; Silva, S.; Henriques, M. Candida glabrata Biofilms: How Far Have We Come? J. Fungi 2017, 3, 11. [Google Scholar] [CrossRef]
- Kumar, K.; Askari, F.; Sahu, M.; Kaur, R. Candida glabrata: A Lot More Than Meets the Eye. Microorganisms 2019, 7, 39. [Google Scholar] [CrossRef]
- Hasan, F.; Xess, I.; Wang, X.; Jain, N.; Fries, B.C. Biofilm formation in clinical Candida isolates and its association with virulence. Microbes Infect. 2009, 11, 753–761. [Google Scholar] [CrossRef]
- Finkel, J.S.; Mitchell, A.P. Genetic control of Candida albicans biofilm development. Nat. Rev. Microbiol. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, K.J.; Klis, F.M. Flocculation, adhesion and biofilm formation in yeasts. Mol. Microbiol. 2006, 60, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.P.; Ho, J.; Naglik, J.R. Candida-Epithelial Interactions. J. Fungi 2018, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.J. Adhesion of Candida species to epithelial surfaces. Crit. Rev. Microbiol. 1987, 15, 27–43. [Google Scholar] [CrossRef]
- Höfs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169. [Google Scholar] [CrossRef]
- de Groot, P.W.J.; Bader, O.; de Boer, A.D.; Weig, M.; Chauhan, N. Adhesins in human fungal pathogens: Glue with plenty of stick. Eukaryot. Cell 2013, 12, 470–481. [Google Scholar] [CrossRef]
- López-Fuentes, E.; Gutiérrez-Escobedo, G.; Timmermans, B.; Van Dijck, P.; De Las Peñas, A.; Castaño, I. Candida glabrata’s Genome Plasticity Confers a Unique Pattern of Expressed Cell Wall Proteins. J. Fungi 2018, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Samaranayake, L.P. Candida glabrata, an emerging fungal pathogen, exhibits superior relative cell surface hydrophobicity and adhesion to denture acrylic surfaces compared with Candida albicans. APMIS 2002, 110, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.T.; Sacristán, B.; Lucio, L.; Blanco, J.; Pérez-Giraldo, C.; Gómez-García, A.C. Cell surface hydrophobicity as an indicator of other virulence factors in Candida albicans. Rev. Iberoam. Micol. 2010, 27, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Hazen, K.C.; Plotkin, B.J.; Klimas, D.M. Influence of growth conditions on cell surface hydrophobicity of Candida albicans and Candida glabrata. Infect. Immunity 1986, 54, 269–271. [Google Scholar]
- Hoyer, L.L.; Green, C.B.; Oh, S.-H.; Zhao, X. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family—A sticky pursuit. Med. Mycol. 2008, 46, 1–15. [Google Scholar] [CrossRef]
- Cormack, B.; Zordan, R. Adhesins in Opportunistic Fungal Pathogens. In Candida and Candidiasis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2012; pp. 243–259. [Google Scholar]
- Wächtler, B.; Wilson, D.; Haedicke, K.; Dalle, F.; Hube, B. From attachment to damage: Defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS ONE 2011, 6, e17046. [Google Scholar] [CrossRef]
- Nobile, C.J.; Schneider, H.A.; Nett, J.E.; Sheppard, D.C.; Filler, S.G.; Andes, D.R.; Mitchell, A.P. Complementary adhesin function in C. albicans biofilm formation. Curr. Biol. 2008, 18, 1017–1024. [Google Scholar] [CrossRef]
- Younes, S.; Bahnan, W.; Dimassi, H.I.; Khalaf, R.A. The Candida albicans Hwp2 is necessary for proper adhesion, biofilm formation and oxidative stress tolerance. Microbiol. Res. 2011, 166, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Svarovsky, M.J.; Karlsson, A.J.; Wagner, J.P.; Marchillo, K.; Oshel, P.; Andes, D.; Palecek, S.P. Eap1p, an adhesin that mediates Candida albicans biofilm formation in vitro and in vivo. Eukaryot. Cell 2007, 6, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Palecek, S.P. EAP1, a Candida albicans gene involved in binding human epithelial cells. Eukaryot. Cell 2003, 2, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Semlali, A.; Killer, K.; Alanazi, H.; Chmielewski, W.; Rouabhia, M. Cigarette smoke condensate increases C. albicans adhesion, growth, biofilm formation, and EAP1, HWP1 and SAP2 gene expression. BMC Microbiol. 2014, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Ghori, N.; Falkow, S. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 1999, 285, 578–582. [Google Scholar] [CrossRef]
- Halliwell, S.C.; Smith, M.C.A.; Muston, P.; Holland, S.L.; Avery, S.V. Heterogeneous expression of the virulence-related adhesin epa1 between individual cells and strains of the pathogen Candida glabrata. Eukaryot. Cell 2012, 11, 141–150. [Google Scholar] [CrossRef]
- De Las Peñas, A.; Pan, S.J.; Castaño, I.; Alder, J.; Cregg, R.; Cormack, B.P. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 2003, 17, 2245–2258. [Google Scholar] [CrossRef]
- Domergue, R.; Castaño, I.; De Las Peñas, A.; Zupancic, M.; Lockatell, V.; Hebel, J.R.; Johnson, D.; Cormack, B.P. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 2005, 308, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Mogensen, E.; d’Enfert, C.; Janbon, G. New regulators of biofilm development in Candida glabrata. Res. Microbiol. 2012, 163, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Iraqui, I.; Garcia-Sanchez, S.; Aubert, S.; Dromer, F.; Ghigo, J.M.; D’Enfert, C.; Janbon, G. The Yak1p kinase controls expression of adhesins and biofilm formation in Candida glabrata in a Sir4p-dependent pathway. Mol. Microbiol. 2005, 55, 1259–1271. [Google Scholar] [CrossRef]
- Kraneveld, E.A.; de Soet, J.J.; Deng, D.M.; Dekker, H.L.; de Koster, C.G.; Klis, F.M.; Crielaard, W.; de Groot, P.W.J. Identification and Differential Gene Expression of Adhesin-Like Wall Proteins in Candida glabrata Biofilms. Mycopathologia 2011, 172, 415–427. [Google Scholar] [CrossRef]
- Gómez-Molero, E.; de Boer, A.D.; Dekker, H.L.; Moreno-Martínez, A.; Kraneveld, E.A.; Chauhan, N.; Weig, M.; de Soet, J.J.; de Koster, C.G.; et al. Proteomic analysis of hyperadhesive Candida glabrata clinical isolates reveals a core wall proteome and differential incorporation of adhesins. FEMS Yeast Res. 2015, 15, fov098. [Google Scholar] [CrossRef]
- Leiva-Peláez, O.; Gutiérrez-Escobedo, G.; López-Fuentes, E.; Cruz-Mora, J.; De Las Peñas, A.; Castaño, I. Molecular characterization of the silencing complex SIR in Candida glabrata hyperadherent clinical isolates. Fungal Genet. Biol. 2018, 118, 21–31. [Google Scholar] [CrossRef]
- Ene, I.V.; Adya, A.K.; Wehmeier, S.; Brand, A.C.; MacCallum, D.M.; Gow, N.A.R.; Brown, A.J.P. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell. Microbiol. 2012, 14, 1319–1335. [Google Scholar] [CrossRef] [Green Version]
- Van Ende, M.; Wijnants, S.; Van Dijck, P. Sugar Sensing and Signaling in Candida albicans and Candida glabrata. Front. Microbiol. 2019, 10, 99. [Google Scholar] [CrossRef]
- Vale-Silva, L.; Beaudoing, E.; Tran, V.D.T.; Sanglard, D. Comparative Genomics of Two Sequential Candida glabrata Clinical Isolates. G3 Genes Genomes Genet. 2017, 7, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Wang, C.; Tian, Y.; Dong, D.; Jiang, C.; Mao, E.; Peng, Y. CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 2018, 61, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.A.; Moeckli, B.; Torelli, R.; Posteraro, B.; Sanguinetti, M.; Sanglard, D. Upregulation of the Adhesin Gene EPA1 Mediated by PDR1 in Candida glabrata Leads to Enhanced Host Colonization. mSphere 2016, 1, e00065-15. [Google Scholar] [CrossRef]
- Filler, S.G. Candida-host cell receptor-ligand interactions. Curr. Opin. Microbiol. 2006, 9, 333–339. [Google Scholar] [CrossRef]
- Salazar, S.B.; Wang, C.; Münsterkötter, M.; Okamoto, M.; Takahashi-Nakaguchi, A.; Chibana, H.; Lopes, M.M.; Güldener, U.; Butler, G.; Mira, N.P. Comparative genomic and transcriptomic analyses unveil novel features of azole resistance and adaptation to the human host in Candida glabrata. FEMS Yeast Res. 2018, 18, fox079. [Google Scholar] [CrossRef]
- Vale-Silva, L.; Ischer, F.; Leibundgut-Landmann, S.; Sanglard, D. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect. Immunity 2013, 81, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Vasconcelos, S.S.; Fiorini, A.; Zanni, P.D.; Bonfim-Mendonça, P.; Godoy, J.R.; Almeida-Apolonio, A.A.; Consolaro, M.E.L.; Svidzinski, T.I.E. Emergence of Candida glabrata in vulvovaginal candidiasis should be attributed to selective pressure or virulence ability? Arch. Gynecol. Obstet. 2017, 296, 519–526. [Google Scholar] [CrossRef]
- Fox, E.P.; Bui, C.K.; Nett, J.E.; Hartooni, N.; Mui, M.C.; Andes, D.R.; Nobile, C.J.; Johnson, A.D. An expanded regulatory network temporally controls Candida albicans biofilm formation. Mol. Microbiol. 2015, 96, 1226–1239. [Google Scholar] [CrossRef]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A Recently Evolved Transcriptional Network Controls Biofilm Development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef]
- Nobile, C.J.; Andes, D.R.; Nett, J.E.; Smith, F.J.; Yue, F.; Phan, Q.; Edwards, J.E.; Filler, S.G.; Mitchell, A.P. Critical Role of Bcr1-Dependent Adhesins in C. albicans Biofilm Formation In Vitro and In Vivo. PLoS Pathog. 2006, 2, 0636–0649. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Nett, J.E.; Andes, D.R.; Mitchell, A.P. Function of Candida albicans adhesin Hwp1 in biofilm formation. Eukaryot. Cell 2006, 5, 1604–1610. [Google Scholar] [CrossRef]
- Finkel, J.S.; Xu, W.; Huang, D.; Hill, E.M.; Desai, J.V.; Woolford, C.A.; Nett, J.E.; Taff, H.; Norice, C.T.; Andes, D.R.; et al. Portrait of Candida albicans adherence regulators. PLoS Pathog. 2012, 8, 1–14. [Google Scholar] [CrossRef]
- Silva-Dias, A.; Miranda, I.M.; Branco, J.; Monteiro-Soares, M.; Pina-Vaz, C.; Rodrigues, A.G. Adhesion, biofilm formation, cell surface hydrophobicity, and antifungal planktonic susceptibility: Relationship among Candida spp. Front. Microbiol. 2015, 6, 205. [Google Scholar] [CrossRef]
- Bendel, C.M. Colonization and epithelial adhesion in the pathogenesis of neonatal candidiasis. Semin. Perinatol. 2003, 27, 357–364. [Google Scholar] [CrossRef]
- Lo, H.J.; Köhler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous C. albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef]
- Nobile, C.J.; Mitchell, A.P. Genetics and genomics of Candida albicans biofilm formation. Cell. Microbiol. 2006, 8, 1382–1391. [Google Scholar] [CrossRef]
- Douglas, L.J. Candida biofilms and their role in infection. Trends Microbiol. 2003, 11, 30–36. [Google Scholar] [CrossRef]
- Ramage, G.; Martínez, J.P.; López-Ribot, J.L. Candida biofilms on implanted biomaterials: A clinically significant problem. FEMS Yeast Res. 2006, 6, 979–986. [Google Scholar] [CrossRef]
- Rodríguez-cerdeira, C.; Gregorio, M.C.; Molares-vila, A.; López-barcenas, A.; Fabbrocini, G.; Bardhi, B.; Sinani, A.; Sánchez-blanco, E.; Arenas-guzmán, R.; Hernandez-castro, R. Biofilms and vulvovaginal candidiasis. Colloids Surf. B Biointerfaces 2019, 174, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Mukherjee, P.K. Candida Biofilms: Development, Architecture, and Resistance. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Kucharíková, S.; Neirinck, B.; Sharma, N.; Vleugels, J.; Lagrou, K.; Van Dijck, P. In vivo Candida glabrata biofilm development on foreign bodies in a rat subcutaneous model. J. Antimicrob. Chemother. 2015, 70, 846–856. [Google Scholar] [CrossRef]
- Silva, S.; Henriques, M.; Martins, A.; Oliveira, R.; Williams, D.; Azeredo, J. Biofilms of non-Candida albicans Candida species: Quantification, structure and matrix composition. Med. Mycol. 2009, 47, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, C.J.; Silva, W.J.; Jin, L.J.; Samaranayake, Y.H.; Samaranayake, L.P. Architectural analysis, viability assessment and growth kinetics of Candida albicans and Candida glabrata biofilms. Arch. Oral Biol. 2009, 54, 1052–1060. [Google Scholar] [CrossRef]
- Ramage, G.; VandeWalle, K.; Wickes, B.L.; López-Ribot, J.L. Characteristics of biofilm formation by Candida albicans. Rev. Iberoam. Micol. 2001, 18, 163–170. [Google Scholar]
- Hawser, S.P.; Douglas, L.J. Biofilm formation by Candida species on the surface of catheter materials in vitro. Infect. Immunity 1994, 62, 915–921. [Google Scholar]
- Lewis, R.E.; Lo, H.; Raad, I.I.; Kontoyiannis, D.P. Lack of Catheter Infection by the efg1/efg1 cph1/cph1 Double-Null Mutant, a Candida albicans Strain That Is Defective in Filamentous Growth. Antimicrob. Agents Chemother. 2002, 46, 1153–1155. [Google Scholar] [CrossRef] [PubMed]
- Ramage, G.; Vandewalle, K.; López-Ribot, J.L.; Wickes, B.L. The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol. Lett. 2002, 214, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Thein, Z.M.; Samaranayake, Y.H.; Samaranayake, L.P. In vitro biofilm formation of Candida albicans and non-albicans Candida species under dynamic and anaerobic conditions. Arch. Oral Biol. 2007, 52, 761–767. [Google Scholar] [CrossRef]
- Fonseca, E.; Silva, S.; Rodrigues, C.F.; Alves, C.T.; Azeredo, J.; Henriques, M. Effects of fluconazole on Candida glabrata biofilms and its relationship with ABC transporter gene expression. Biofouling 2014, 30, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Zarnowski, R.; Westler, W.M.; de Lacmbouh, G.A.; Marita, J.M.; Bothe, J.R.; Bernhardt, J.; Lounes-Hadj Sahraoui, A.; Fontaine, J.; Sanchez, H.; Hatfield, R.D.; et al. Novel entries in a fungal biofilm matrix encyclopedia. MBio 2014, 5, e01333-14. [Google Scholar] [CrossRef] [PubMed]
- Al-Fattani, M.A.; Douglas, L.J. Biofilm matrix of Candida albicans and Candida tropicalis: Chemical composition and role in drug resistance. J. Med. Microbiol. 2006, 55, 999–1008. [Google Scholar] [CrossRef]
- Susewind, S.; Lang, R.; Hahnel, S. Biofilm formation and Candida albicans morphology on the surface of denture base materials. Mycoses 2015, 58, 719–727. [Google Scholar] [CrossRef]
- Chandra, J.; Patel, J.P.; Li, J.; Zhou, G.; Mukherjee, P.K.; Mccormick, T.S.; Anderson, J.M.; Ghannoum, M. A Modification of Surface Properties of Biomaterials Influences the Ability of Candida albicans to Form Biofilms. Appl. Environ. Microbiol. 2005, 71, 8795–8801. [Google Scholar] [CrossRef]
- Estivill, D.; Arias, A.; Torres-Lana, A.; Carrillo-Muñoz, A.J.; Arévalo, M.P. Biofilm formation by five species of Candida on three clinical materials. J. Microbiol. Methods 2011, 86, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Mutluay, M.M.; Oǧuz, S.; Ørstavik, D.; Fløystrand, F.; Doǧan, A.; Söderling, E.; Närhi, T.; Olsen, I. Experiments on in vivo biofilm formation and in vitro adhesion of Candida species on polysiloxane liners. Gerodontology 2010, 27, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Samaranayake, L.P.; Samaranayake, Y.; Yip, H.K. Biofilm formation of Candida albicans is variably affected by saliva and dietary sugars. Arch. Oral Biol. 2004, 49, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Shin, J.H.; Jung, S.I.; Park, K.H.; Cho, D.; Kee, S.J.; Shin, M.G.; Suh, S.P.; Ryang, D.W. Species-specific differences in the susceptibilities of biofilms formed by Candida bloodstream isolates to echinocandin antifungals. Antimicrob. Agents Chemother. 2007, 51, 1520–1523. [Google Scholar] [CrossRef]
- Valentín, A.; Cantó, E.; Pemán, J.; Martínez, J.P. Voriconazole inhibits biofilm formation in different species of the genus Candida. J. Antimicrob. Chemother. 2012, 67, 2418–2423. [Google Scholar] [CrossRef] [PubMed]
- Kucharíková, S.; Tournu, H.; Lagrou, K.; van Dijck, P.; Bujdáková, H. Detailed comparison of Candida albicans and Candida glabrata biofilms under different conditions and their susceptibility to caspofungin and anidulafungin. J. Med. Microbiol. 2011, 60, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Taff, H.T.; Mitchell, K.F.; Edward, J.A.; Andes, D.R. Mechanisms of Candida biofilm drug resistance. Future Microbiol. 2013, 8, 1325–1337. [Google Scholar] [CrossRef]
- Ramage, G.; Bachmann, S.; Patterson, T.F.; Wickes, B.L.; López-ribot, J.L. Investigation of multidrug efflux pumps in relation to fluconazole resistance in Candida albicans biofilms. J. Antimicrob. Chemother. 2002, 49, 973–980. [Google Scholar] [CrossRef]
- Vediyappan, G.; Rossignol, T.; D’Enfert, C. Interaction of Candida albicans biofilms with antifungals: Transcriptional response and binding of antifungals to beta-glucans. Antimicrob. Agents Chemother. 2010, 54, 2096–2111. [Google Scholar] [CrossRef]
- Rodrigues, C.F.; Silva, S.; Azeredo, J.; Henriques, M. Candida glabrata’s recurrent infections: Biofilm formation during Amphotericin B treatment. Lett. Appl. Microbiol. 2016, 63, 77–81. [Google Scholar] [CrossRef]
- Johnson, C.J.; Cabezas-Olcoz, J.; Kernien, J.F.; Wang, S.X.; Beebe, D.J.; Huttenlocher, A.; Ansari, H.; Nett, J.E. The Extracellular Matrix of Candida albicans Biofilms Impairs Formation of Neutrophil Extracellular Traps. PLoS Pathog. 2016, 12, 1–23. [Google Scholar] [CrossRef]
- Xie, Z.; Thompson, A.; Sobue, T.; Kashleva, H.; Xu, H.; Vasilakos, J.; Dongari-Bagtzoglou, A. Candida albicans biofilms do not trigger reactive oxygen species and evade neutrophil killing. J. Infect. Dis. 2012, 206, 1936–1945. [Google Scholar] [CrossRef]
- Lohse, M.B.; Gulati, M.; Johnson, A.D.; Nobile, C.J. Development and regulation of single-and multi-species Candida albicans biofilms. Nat. Rev. Microbiol. 2018, 16, 19–31. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Brown, G.D.; Gordon, S. Immune recognition. A new receptor for beta-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef]
- Li, M.; Chen, Q.; Tang, R.; Shen, Y.; Liu, W. Da The expression of β-defensin-2, 3 and LL-37 induced by Candida albicans phospholipomannan in human keratinocytes. J. Dermatol. Sci. 2011, 61, 72–75. [Google Scholar] [CrossRef]
- Tomalka, J.; Azodi, E.; Narra, H.P.; Patel, K.; O’Neill, S.; Cardwell, C.; Hall, B.A.; Wilson, J.M.; Hise, A.G. β-Defensin 1 Plays a Role in Acute Mucosal Defense against Candida albicans. J. Immunol. 2015, 194, 1788–1795. [Google Scholar] [CrossRef]
- Järvå, M.; Phan, T.K.; Lay, F.T.; Caria, S.; Kvansakul, M.; Hulett, M.D. Human β-defensin 2 kills Candida albicans through phosphatidylinositol 4,5-bisphosphate-mediated membrane permeabilization. Sci. Adv. 2018, 4, eaat0979. [Google Scholar] [CrossRef]
- Hardison, S.E.; Brown, G.D. C-type lectin receptors orchestrate antifungal immunity. Nat. Immunol. 2012, 13, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Maródi, L. Innate immune mechanisms for recognition and uptake of Candida species. Trends Immunol. 2010, 31, 346–353. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Joosten, L.A.B.; Kullberg, B.-J.; Netea, M.G. Interplay between Candida albicans and the mammalian innate host defense. Infect. Immunity 2012, 80, 1304–1313. [Google Scholar] [CrossRef]
- Gilbert, A.S.; Wheeler, R.T.; May, R.C. Fungal Pathogens: Survival and Replication within Macrophages. Cold Spring Harb. Perspect. Med. 2014, 5, a019661. [Google Scholar] [CrossRef] [Green Version]
- Free, S.J. Fungal Cell Wall Organization and Biosynthesis; Academic Press: Cambridge, MA, USA, 2013; pp. 33–82. [Google Scholar]
- Graus, M.S.; Wester, M.J.; Lowman, D.W.; Williams, D.L.; Kruppa, M.D.; Martinez, C.M.; Young, J.M.; Pappas, H.C.; Lidke, K.A.; Neumann, A.K. Mannan Molecular Substructures Control Nanoscale Glucan Exposure in Candida. Cell Rep. 2018, 24, 2432–2442. [Google Scholar] [CrossRef]
- Ballou, E.R.; Avelar, G.M.; Childers, D.S.; Mackie, J.; Bain, J.M.; Wagener, J.; Kastora, S.L.; Panea, M.D.; Hardison, S.E.; Walker, L.A.; et al. Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat. Microbiol. 2016, 2, 16238. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Lopes, J.P.; Stylianou, M.; Backman, E.; Holmberg, S.; Jass, J.; Claesson, R.; Urban, C.F. Evasion of Immune Surveillance in Low Oxygen Environments Enhances Candida albicans Virulence. MBio 2018, 9, e02120-18. [Google Scholar] [CrossRef]
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.S.; Larcombe, D.E.; Netea, M.G.; Shekhova, E.; Munro, C.A.; Brown, G.D.; Erwig, L.P.; et al. Hypoxia Promotes Immune Evasion by Triggering β-Glucan Masking on the Candida albicans Cell Surface via Mitochondrial and cAMP-Protein Kinase A Signaling. MBio 2018, 9, e01318-18. [Google Scholar] [CrossRef]
- Ene, I.V.; Cheng, S.-C.; Netea, M.G.; Brown, A.J.P. Growth of Candida albicans cells on the physiologically relevant carbon source lactate affects their recognition and phagocytosis by immune cells. Infect. Immunity 2013, 81, 238–248. [Google Scholar] [CrossRef]
- Klis, F.M.; de Groot, P.; Hellingwerf, K. Molecular organization of the cell wall of Candida albicans. Med. Mycol. 2001, 39 (Suppl. 1), 1–8. [Google Scholar] [CrossRef]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A.R. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, J.; Victoria Elorza, M.; ValentÃ-n, E.; Sentandreu, R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006, 6, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.T.; Kombe, D.; Agarwala, S.D.; Fink, G.R. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. 2008, 4, e1000227. [Google Scholar] [CrossRef] [PubMed]
- Hopke, A.; Nicke, N.; Hidu, E.E.; Degani, G.; Popolo, L.; Wheeler, R.T. Neutrophil Attack Triggers Extracellular Trap-Dependent Candida Cell Wall Remodeling and Altered Immune Recognition. PLoS Pathog. 2016, 12, e1005644. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, S.L.; Sorsby, E.; Mahtey, N.; Kumwenda, P.; Lenardon, M.D.; Brown, I.; Ballou, E.R.; MacCallum, D.M.; Hall, R.A. Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog. 2017, 13, e1006403. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.T.; Fink, G.R. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A.; Bates, S.; Lenardon, M.D.; MacCallum, D.M.; Wagener, J.; Lowman, D.W.; Kruppa, M.D.; Williams, D.L.; Odds, F.C.; Brown, A.J.P.; et al. The Mnn2 Mannosyltransferase Family Modulates Mannoprotein Fibril Length, Immune Recognition and Virulence of Candida albicans. PLoS Pathog. 2013, 9, e1003276. [Google Scholar] [CrossRef]
- Rouabhia, M.; Schaller, M.; Corbucci, C.; Vecchiarelli, A.; Prill, S.K.-H.; Giasson, L.; Ernst, J.F. Virulence of the fungal pathogen Candida albicans requires the five isoforms of protein mannosyltransferases. Infect. Immunity 2005, 73, 4571–4580. [Google Scholar] [CrossRef] [PubMed]
- West, L.; Lowman, D.W.; Mora-Montes, H.M.; Grubb, S.; Murdoch, C.; Thornhill, M.H.; Gow, N.A.R.; Williams, D.; Haynes, K. Differential virulence of Candida glabrata glycosylation mutants. J. Biol. Chem. 2013, 288, 22006–22018. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Zou, Z.; Shen, H.; Shen, S.S.; Miao, Q.; Huang, X.; Liu, W.; Li, L.P.; Chen, S.M.; Yan, L.; et al. Mnn10 Maintains Pathogenicity in Candida albicans by Extending α-1,6-Mannose Backbone to Evade Host Dectin-1 Mediated Antifungal Immunity. PLoS Pathog. 2016, 12, e1005617. [Google Scholar] [CrossRef]
- Keppler-Ross, S.; Douglas, L.; Konopka, J.B.; Dean, N. Recognition of Yeast by Murine Macrophages Requires Mannan but Not Glucan. Eukaryot. Cell 2010, 9, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Shen, H.; Zhang, T.; Huang, X.; Liu, X.Q.; Guo, S.Y.; Zhao, J.J.; Wang, C.F.; Yan, L.; Xu, G.T.; et al. Dectin-1 plays an important role in host defense against systemic Candida glabrata infection. Virulence 2017, 8, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Ifrim, D.C.; Bain, J.M.; Reid, D.M.; Oosting, M.; Verschueren, I.; Gow, N.A.R.; van Krieken, J.H.; Brown, G.D.; Kullberg, B.-J.; Joosten, L.A.B.; et al. Role of Dectin-2 for Host Defense against Systemic Infection with Candida glabrata. Infect. Immunity 2014, 82, 1064–1073. [Google Scholar] [CrossRef]
- Jacobsen, I.D.; Brunke, S.; Seider, K.; Schwarzmüller, T.; Firon, A.; d’Enfért, C.; Kuchler, K.; Hube, B. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect. Immunity 2010, 78, 1066–1077. [Google Scholar] [CrossRef]
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2008, 6, 132–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poltermann, S.; Kunert, A.; von der Heide, M.; Eck, R.; Hartmann, A.; Zipfel, P.F. Gpm1p is a factor H-, FHL-1-, and plasminogen-binding surface protein of Candida albicans. J. Biol. Chem. 2007, 282, 37537–37544. [Google Scholar] [CrossRef]
- Vogl, G.; Lesiak, I.; Jensen, D.B.; Perkhofer, S.; Eck, R.; Speth, C.; Lass-Flörl, C.; Zipfel, P.F.; Blom, A.M.; Dierich, M.P.; et al. Immune evasion by acquisition of complement inhibitors: The mould Aspergillus binds both factor H and C4b binding protein. Mol. Immunol. 2008, 45, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Poltermann, S.; Kunert, A.; Rupp, S.; Zipfel, P.F. Immune evasion of the human pathogenic yeast Candida albicans: Pra1 is a Factor H, FHL-1 and plasminogen binding surface protein. Mol. Immunol. 2009, 47, 541–550. [Google Scholar] [CrossRef]
- Kenno, S.; Speth, C.; Rambach, G.; Binder, U.; Chatterjee, S.; Caramalho, R.; Haas, H.; Lass-Flörl, C.; Shaughnessy, J.; Ram, S.; et al. Candida albicans Factor H Binding Molecule Hgt1p—A Low Glucose-Induced Transmembrane Protein Is Trafficked to the Cell Wall and Impairs Phagocytosis and Killing by Human Neutrophils. Front. Microbiol. 2018, 9, 3319. [Google Scholar] [CrossRef]
- Staib, F. Serum-proteins as nitrogen source for yeastlike fungi. Sabouraudia 1965, 4, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Gropp, K.; Schild, L.; Schindler, S.; Hube, B.; Zipfel, P.F.; Skerka, C. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol. Immunol. 2009, 47, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Meiller, T.F.; Hube, B.; Schild, L.; Shirtliff, M.E.; Scheper, M.A.; Winkler, R.; Ton, A.; Jabra-Rizk, M.A. A novel immune evasion strategy of Candida albicans: Proteolytic cleavage of a salivary antimicrobial peptide. PLoS ONE 2009, 4, e5039. [Google Scholar] [CrossRef]
- Svoboda, E.; Schneider, A.E.; Sándor, N.; Lermann, U.; Staib, P.; Kremlitzka, M.; Bajtay, Z.; Barz, D.; Erdei, A.; Józsi, M. Secreted aspartic protease 2 of Candida albicans inactivates factor H and the macrophage factor H-receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18). Immunol. Lett. 2015, 168, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hallström, T.; Singh, B.; Kraiczy, P.; Hammerschmidt, S.; Skerka, C.; Zipfel, P.F.; Riesbeck, K. Conserved Patterns of Microbial Immune Escape: Pathogenic Microbes of Diverse Origin Target the Human Terminal Complement Inhibitor Vitronectin via a Single Common Motif. PLoS ONE 2016, 11, e0147709. [Google Scholar] [CrossRef]
- Luo, S.; Dasari, P.; Reiher, N.; Hartmann, A.; Jacksch, S.; Wende, E.; Barz, D.; Niemiec, M.J.; Jacobsen, I.; Beyersdorf, N.; et al. The secreted Candida albicans protein Pra1 disrupts host defense by broadly targeting and blocking complement C3 and C3 activation fragments. Mol. Immunol. 2018, 93, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, A.; Dasari, P.; Werner, S.; Hughes, T.R.; Song, W.-C.; Hortschansky, P.; Brakhage, A.A.; Hünig, T.; Zipfel, P.F.; Beyersdorf, N. Direct Binding of the pH-Regulated Protein 1 (Pra1) from Candida albicans Inhibits Cytokine Secretion by Mouse CD4+ T Cells. Front. Microbiol. 2017, 8, 844. [Google Scholar] [CrossRef]
- Vylkova, S.; Carman, A.J.; Danhof, H.A.; Collette, J.R.; Zhou, H.; Lorenz, M.C. The fungal pathogen Candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. MBio 2011, 2, e00055-11. [Google Scholar] [CrossRef]
- Brown, A.J.P.; Brown, G.D.; Netea, M.G.; Gow, N.A.R. Metabolism impacts upon Candida immunogenicity and pathogenicity at multiple levels. Trends Microbiol. 2014, 22, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M.C.; Fink, G.R. The glyoxylate cycle is required for fungal virulence. Nature 2001, 412, 83. [Google Scholar] [CrossRef]
- Fernández-Arenas, E.; Bleck, C.K.E.; Nombela, C.; Gil, C.; Griffiths, G.; Diez-Orejas, R. Candida albicans actively modulates intracellular membrane trafficking in mouse macrophage phagosomes. Cell. Microbiol. 2009, 11, 560–589. [Google Scholar] [CrossRef]
- Kasper, L.; Seider, K.; Gerwien, F.; Allert, S.; Brunke, S.; Schwarzmüller, T.; Ames, L.; Zubiria-Barrera, C.; Mansour, M.K.; Becken, U.; et al. Identification of Candida glabrata genes involved in pH modulation and modification of the phagosomal environment in macrophages. PLoS ONE 2014, 9, e96015. [Google Scholar] [CrossRef]
- Vylkova, S.; Lorenz, M.C. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog. 2014, 10, e1003995. [Google Scholar] [CrossRef]
- Rai, M.N.; Balusu, S.; Gorityala, N.; Dandu, L.; Kaur, R. Functional Genomic Analysis of Candida glabrata-Macrophage Interaction: Role of Chromatin Remodeling in Virulence. PLoS Pathog. 2012, 8, e1002863. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.Y.; Ho, K.L.; Cheah, Y.K.; Ng, T.S.; Sandai, D.; Brown, A.J.P.; Than, L.T.L. Glyoxylate cycle gene ICL1 is essential for the metabolic flexibility and virulence of Candida glabrata. Sci. Rep. 2019, 9, 2843. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Naseem, S.; Sharma, S.; Konopka, J.B. Flavodoxin-Like Proteins Protect Candida albicans from Oxidative Stress and Promote Virulence. PLoS Pathog. 2015, 11, e1005147. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.; MacCallum, D.M.; Brown, G.D.; Gow, N.A.R. Candida albicans Chitin Increases Arginase-1 Activity in Human Macrophages, with an Impact on Macrophage Antimicrobial Functions. MBio 2017, 8, e01820-16. [Google Scholar] [CrossRef]
- Ghosh, S.; Navarathna, D.H.M.L.P.; Roberts, D.D.; Cooper, J.T.; Atkin, A.L.; Petro, T.M.; Nickerson, K.W. Arginine-induced germ tube formation in Candida albicans is essential for escape from murine macrophage line RAW 264.7. Infect. Immunity 2009, 77, 1596–1605. [Google Scholar] [CrossRef]
- Danhof, H.A.; Lorenz, M.C. The Candida albicans ATO Gene Family Promotes Neutralization of the Macrophage Phagolysosome. Infect. Immunity 2015, 83, 4416–4426. [Google Scholar] [CrossRef]
- Miramón, P.; Lorenz, M.C. The SPS amino acid sensor mediates nutrient acquisition and immune evasion in Candida albicans. Cell. Microbiol. 2016, 18, 1611–1624. [Google Scholar] [CrossRef]
- Cuéllar-Cruz, M.; Briones-Martin-del-Campo, M.; Cañas-Villamar, I.; Montalvo-Arredondo, J.; Riego-Ruiz, L.; Castaño, I.; De Las Peñas, A. High Resistance to Oxidative Stress in the Fungal Pathogen Candida glabrata Is Mediated by a Single Catalase, Cta1p, and Is Controlled by the Transcription Factors Yap1p, Skn7p, Msn2p, and Msn4p. Eukaryot. Cell 2008, 7, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Tsai, H.-F.; Myers, T.G.; Bennett, J.E. Transcriptional Profiling of Candida glabrata during Phagocytosis by Neutrophils and in the Infected Mouse Spleen. Infect. Immunity 2013, 81, 1325–1333. [Google Scholar] [CrossRef]
- Mahl, C.D.; Behling, C.S.; Hackenhaar, F.S.; de Carvalho e Silva, M.N.; Putti, J.; Salomon, T.B.; Alves, S.H.; Fuentefria, A.; Benfato, M.S. Induction of ROS generation by fluconazole in Candida glabrata: Activation of antioxidant enzymes and oxidative DNA damage. Diagn. Microbiol. Infect. Dis. 2015, 82, 203–208. [Google Scholar] [CrossRef]
- Seider, K.; Gerwien, F.; Kasper, L.; Allert, S.; Brunke, S.; Jablonowski, N.; Schwarzmüller, T.; Barz, D.; Rupp, S.; Kuchler, K.; et al. Immune Evasion, Stress Resistance, and Efficient Nutrient Acquisition Are Crucial for Intracellular Survival of Candida glabrata within Macrophages. Eukaryot. Cell 2014, 13, 170–183. [Google Scholar] [CrossRef]
- Nevitt, T.; Thiele, D.J. Host Iron Withholding Demands Siderophore Utilization for Candida glabrata to Survive Macrophage Killing. PLoS Pathog. 2011, 7, e1001322. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Suneetha, K.J.; Kaur, R. A systematic analysis reveals an essential role for high-affinity iron uptake system, haemolysin and CFEM domain-containing protein in iron homoeostasis and virulence in Candida glabrata. Biochem. J. 2014, 463, 103–114. [Google Scholar] [CrossRef]
- Sharma, V.; Purushotham, R.; Kaur, R. The Phosphoinositide 3-Kinase Regulates Retrograde Trafficking of the Iron Permease CgFtr1 and Iron Homeostasis in Candida glabrata. J. Biol. Chem. 2016, 291, 24715–24734. [Google Scholar] [CrossRef]
- Wellington, M.; Koselny, K.; Sutterwala, F.S.; Krysan, D.J. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot. Cell 2014, 13, 329–340. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Schroder, K. Antimicrobial functions of inflammasomes. Curr. Opin. Microbiol. 2013, 16, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Tucey, T.M.; Verma, J.; Harrison, P.F.; Snelgrove, S.L.; Lo, T.L.; Scherer, A.K.; Barugahare, A.A.; Powell, D.R.; Wheeler, R.T.; Hickey, M.J.; et al. Glucose Homeostasis Is Important for Immune Cell Viability during Candida Challenge and Host Survival of Systemic Fungal Infection. Cell Metab. 2018, 27, 988–1006.e7. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.; König, A.; Koenig, P.-A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef]
- Rogiers, O.; Frising, U.C.; Kucharíková, S.; Jabra-Rizk, M.A.; van Loo, G.; Van Dijck, P.; Wullaert, A. Candidalysin Crucially Contributes to Nlrp3 Inflammasome Activation by Candida albicans Hyphae. MBio 2019, 10, e02221-18. [Google Scholar] [CrossRef]
- Hargarten, J.C.; Moore, T.C.; Petro, T.M.; Nickerson, K.W.; Atkin, A.L. Candida albicans Quorum Sensing Molecules Stimulate Mouse Macrophage Migration. Infect. Immunity 2015, 83, 3857–3864. [Google Scholar] [CrossRef]
- Brunke, S.; Seider, K.; Fischer, D.; Jacobsen, I.D.; Kasper, L.; Jablonowski, N.; Wartenberg, A.; Bader, O.; Enache-Angoulvant, A.; Schaller, M.; et al. One Small Step for a Yeast—Microevolution within Macrophages Renders Candida glabrata Hypervirulent Due to a Single Point Mutation. PLoS Pathog. 2014, 10, e1004478. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galocha, M.; Pais, P.; Cavalheiro, M.; Pereira, D.; Viana, R.; Teixeira, M.C. Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin. Int. J. Mol. Sci. 2019, 20, 2345. https://doi.org/10.3390/ijms20092345
Galocha M, Pais P, Cavalheiro M, Pereira D, Viana R, Teixeira MC. Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin. International Journal of Molecular Sciences. 2019; 20(9):2345. https://doi.org/10.3390/ijms20092345
Chicago/Turabian StyleGalocha, Mónica, Pedro Pais, Mafalda Cavalheiro, Diana Pereira, Romeu Viana, and Miguel C. Teixeira. 2019. "Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin" International Journal of Molecular Sciences 20, no. 9: 2345. https://doi.org/10.3390/ijms20092345
APA StyleGalocha, M., Pais, P., Cavalheiro, M., Pereira, D., Viana, R., & Teixeira, M. C. (2019). Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin. International Journal of Molecular Sciences, 20(9), 2345. https://doi.org/10.3390/ijms20092345