Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics



{kind=link}
Abstract
:1. Information Crisis in Bioscience and Cancer Research
2. Regulation of the Human Genome: Networking by Self-Organisation Is the Second Principle of Genome Regulation after WATSON-CRICK Complementarity
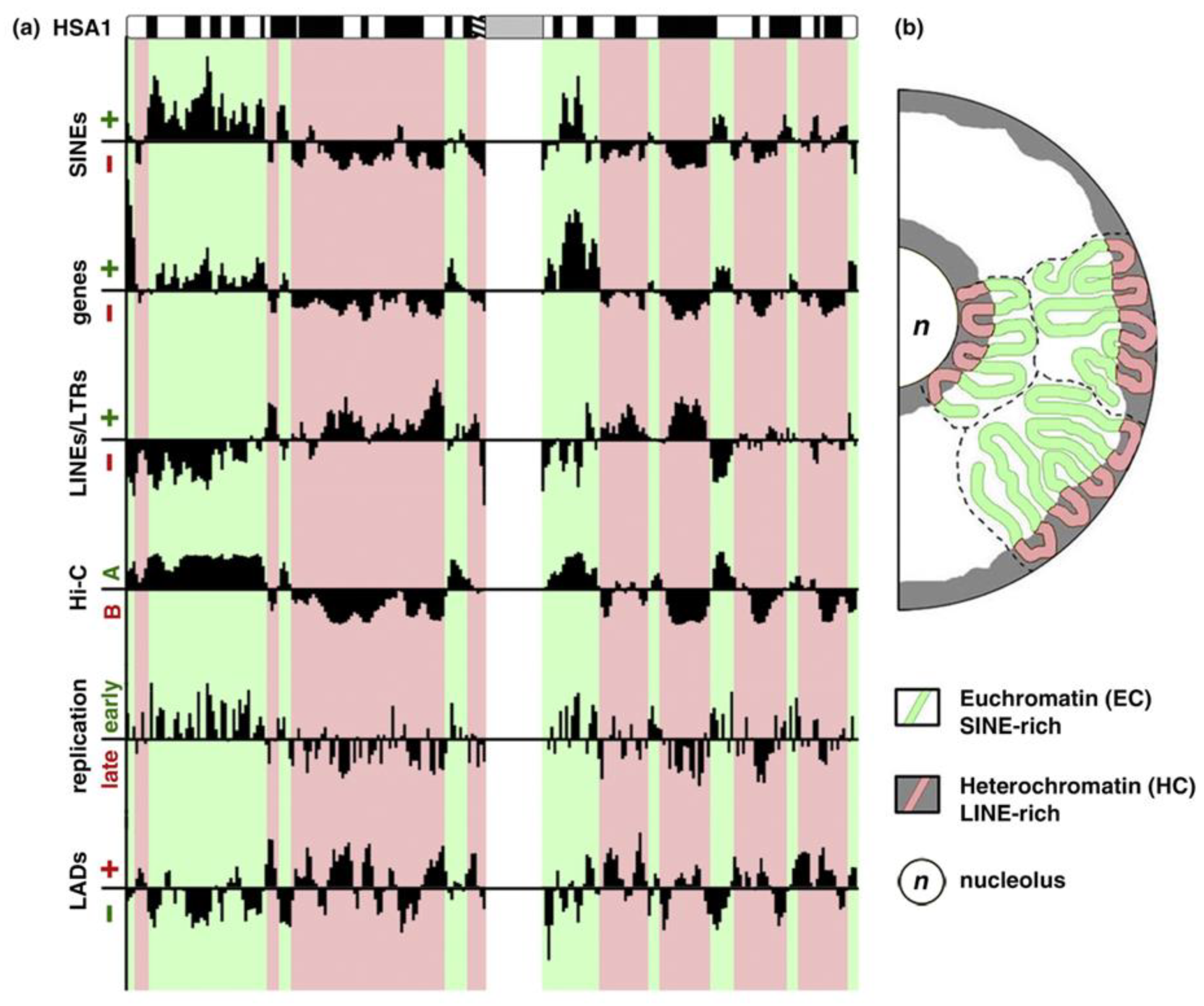
3. The Genome “Maps” of Positional Information Need Phase Transitions
4. Differential DNA Replication Timing Translates Temporal Information into Positional Information
5. Deterministic Chaos for Cell Fate Change: Inevitable Heterogeneity and Fluctuations
6. Cancer Cell Treatment Resistance Is Ensured by Deterministic Chaos and Reprogramming to the Embryonic State
7. Cancer Cells Recapitulate the Stress-Adaptive Programs of Unicellulars and Early Metazoans
8. Chaos in Cancer is Akin to Chaos in an Early Embryo Which Serves to Change Cell Fate
9. Reprogramming of Positional Information Can Be Used for Cancer Reversion Therapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Woese, C.R. A new biology for a new century. Microbiol. Mol. Biol. Rev. 2004, 68, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Heng, H.H.; Liu, G.; Alemara, S.; Regan, S.; Armstrong, Z.; Ye, C.J. The Mechanisms of How Genomic Heterogeneity Impacts Bio-Emergent Properties: The Challenges for Precision Medicine. In Embracing Complexity in Health; Sturmberg, J., Ed.; Springer: Cham, Switzerland, 2019; pp. 95–109. [Google Scholar]
- Ioannidis, J.P.A. Why most published research findings are false. PLoS Med. 2005, 2, e124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuzzo, R. Scientific method: Statistical errors. Nature 2014, 506, 150–152. [Google Scholar] [CrossRef] [Green Version]
- Voosen, P. Amid a Sea of False Findings, the NIH Tries Reform. Chron. High. Educ. 2015, 16, 2015. [Google Scholar]
- Bartova, E. Nuclear structure and gene activity in human differentiated cells. J. Struct. Biol. 2002, 139, 72–89. [Google Scholar] [CrossRef]
- Beil, M.; Dürschmied, D.; Paschke, S.; Schreiner, B.; Nolte, U.; Bruel, A.; Irinopoulou, T. Spatial distribution patterns of interphase centromeres during retinoic acid-induced differentiation of promyelocytic leukemia cells. Cytometry 2002, 47, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Kinney, N.A.; Onufriev, A.V.; Sharakhov, I.V. Quantified effects of chromosome-nuclear envelope attachments on 3D organization of chromosomes. Nucleus 2015, 6, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Mayer, R.; Brero, A.; von Hase, J.; Schroeder, T.; Cremer, T.; Dietzel, S. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 2005, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Németh, A.; Längst, G. Genome organization in and around the nucleolus. Trends Genet. 2011, 27, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Zeller, P.; Gasser, S.M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015, 31, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Thanisch, K.; Feodorova, Y. How to rule the nucleus: Divide et impera. Curr. Opin. Cell Biol. 2016, 40, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosom. Res. 2003, 11, 485–502. [Google Scholar] [CrossRef]
- Lima-de-Faria, A. Chromosome gradient and chromosome field in Agapanthus. Chromosoma 1954, 6, 330–370. [Google Scholar] [CrossRef]
- Lima-de-Faria, A. Classification of genes, rearrangements and chromosomes according to the chromosome field. Hereditas 1980, 93, 1–46. [Google Scholar] [CrossRef]
- Nagl, W. Condensed Chromatin: Species-Specificity, Tissue-Specificity, and Cell Cycle-Specificity, as Monitored by Scanning Cytometry. Cell Growth 1982, 171–218. [Google Scholar]
- Nagl, W.; Popp, F.A. A physical (electromagnetic) model of differentiation. 1. Basic considerations. Cytobios 1983, 37, 45–62. [Google Scholar]
- Popp, F.A.; Nagl, W. A physical (electromagnetic) model of differentiation. 2. Applications and examples. Cytobios 1983, 37, 71–83. [Google Scholar]
- Albiez, H.; Cremer, M.; Tiberi, C.; Vecchio, L.; Schermelleh, L.; Dittrich, S.; Küpper, K.; Joffe, B.; Thormeyer, T.; von Hase, J.; et al. Chromatin domains and the interchromatin compartment form structurally defined and functionally interacting nuclear networks. Chromosom. Res. 2006, 14, 707–733. [Google Scholar] [CrossRef] [PubMed]
- Cabianca, D.S.; Gasser, S.M. Spatial segregation of heterochromatin: Uncovering functionality in a multicellular organism. Nucleus 2016, 7, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremer, T.; Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Zhukotsky, A.; Kozlov, A. The chromatin network: Image analysis of differentiating chick embryo chondrocytes. Eur. J. Histochem. 1993, 37, 139–147. [Google Scholar] [PubMed]
- Ērenpreisa, J.; Zhukotsky, A. Interphase genome as the active space: Chromatin dynamics during chick embryo chondrogenesis. Mech. Ageing Dev. 1993, 67, 21–32. [Google Scholar] [CrossRef]
- Kosak, S.T.; Groudine, M. Form follows function: The genomic organization of cellular differentiation. Genes Dev. 2004, 18, 1371–1384. [Google Scholar] [CrossRef] [Green Version]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef]
- Wiblin, A.E.; Cui, W.; Clark, A.J.; Bickmore, W.A. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J. Cell Sci. 2005, 118, 3861–3868. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, K.S.; Li, G.; Poh, H.M.; Quek, Y.L.K.; Sia, Y.Y.; Peh, S.Q.; Mulawadi, F.H.; Lim, J.; Sikic, M.; Menghi, F.; et al. Large-scale functional organization of long-range chromatin interaction networks. Cell Rep. 2012, 2, 1207–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, D.L. Nuclear domains. J. Cell Sci. 2001, 114, 2891–2893. [Google Scholar] [PubMed]
- Haaf, T.; Ward, D.C. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp. Cell Res. 1996, 224, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapakse, I.; Perlman, M.D.; Scalzo, D.; Kooperberg, C.; Groudine, M.; Kosak, S.T. The emergence of lineage-specific chromosomal topologies from coordinate gene regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 6679–6684. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.M.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharm. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [Green Version]
- Karsenti, E. Self-organization in cell biology: A brief history. Nat. Rev. Mol. Cell Biol. 2008, 9, 255–262. [Google Scholar] [CrossRef]
- Soofi, E.S. Capturing the Intangible Concept of Information. J. Am. Stat. Assoc. 1994, 89, 1243–1254. [Google Scholar] [CrossRef]
- Landau, L.D. On the theory of phase transition. Zh Eksp Teor Fiz 1937, 19–32. [Google Scholar] [CrossRef]
- Cavalli, G.; Misteli, T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The concept of self-organization in cellular architecture. J. Cell Biol. 2001, 155, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Beyond the Sequence: Cellular Organization of Genome Function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, A.; Tsuchiya, M.; Yoshikawa, K. Self-Organization of Genome Expression from Embryo to Terminal Cell Fate: Single-Cell Statistical Mechanics of Biological Regulation. Entropy 2017, 20, 13. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, M.; Giuliani, A.; Hashimoto, M.; Erenpreisa, J.; Yoshikawa, K. Self-Organizing Global Gene Expression Regulated through Criticality: Mechanism of the Cell-Fate Change. PLoS ONE 2016, 11, e0167912. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, S. Chapter 5 Gene Regulation Networks: A Theory for Their Global Structure and Behaviors. Curr. Top. Dev. Biol. 1971, 6, 145–182. [Google Scholar]
- Huang, S.; Kauffman, S.A. Complex gene regulatory networks–from Structure to Biological Observables: Cell fate determination gene regulation. Comput. Complex. 2012, 527–560. [Google Scholar]
- Bak, P.; Chen, K. Self-Organized Criticality. Sci. Am. 1991, 264, 46–53. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Csermely, P. Allo-network drugs: Harnessing allostery in cellular networks. Trends Pharm. Sci. 2011, 32, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paola, L.D.; Di Paola, L.; Giuliani, A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015, 31, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J. Two mechanisms of chromatin compaction. Acta Histochem. 1989, 86, 129–135. [Google Scholar] [CrossRef]
- Marcand, S.; Gasser, S.M.; Gilson, E. Chromatin: A sticky silence. Curr. Biol. 1996, 6, 1222–1225. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.L.; Straight, A.F. RNA-mediated regulation of heterochromatin. Curr. Opin. Cell Biol. 2017, 46, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Krigerts, J.; Salmina, K.; Selga, T.; Sorokins, H.; Freivalds, T. Differential staining of peripheral nuclear chromatin with Acridine orange implies an A-form epichromatin conformation of the DNA. Nucleus 2018, 9, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R. The Crowded Nucleus. In International Review of Cell and Molecular Biology; Academic Press: Cambridge, MA, USA, 2014; pp. 15–26. [Google Scholar]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.-L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef] [Green Version]
- Finn, E.H.; Misteli, T. A genome disconnect. Nat. Genet. 2019, 51, 1205–1206. [Google Scholar] [CrossRef]
- Svensson, E.I. On Reciprocal Causation in the Evolutionary Process. Evol. Biol. 2018, 45, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [Green Version]
- Boulos, R.E.; Drillon, G.; Argoul, F.; Arneodo, A.; Audit, B. Structural organization of human replication timing domains. FEBS Lett. 2015, 589, 2944–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Julienne, H.; Zoufir, A.; Audit, B.; Arneodo, A. Human genome replication proceeds through four chromatin states. PLoS Comput. Biol. 2013, 9, e1003233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikova, T.D. Regulation of DNA replication timing. Mol. Biol. 2013, 47, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Pliss, A.; Malyavantham, K.S.; Bhattacharya, S.; Berezney, R. Chromatin dynamics in living cells: Identification of oscillatory motion. J. Cell. Physiol. 2013, 228, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Bornfleth, H.; Edelmann, P.; Zink, D.; Cremer, T.; Cremer, C. Quantitative Motion Analysis of Subchromosomal Foci in Living Cells Using Four-Dimensional Microscopy. Biophys. J. 1999, 77, 2871–2886. [Google Scholar] [CrossRef] [Green Version]
- Oomen, M.E.; Dekker, J. Epigenetic characteristics of the mitotic chromosome in 1D and 3D. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Mirny, L.A. The fractal globule as a model of chromatin architecture in the cell. Chromosom. Res. 2011, 19, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B.; Bohinc, K. Condensed DNA: Condensing the concepts. Prog. Biophys. Mol. Biol. 2011, 105, 208–222. [Google Scholar] [CrossRef]
- Hübner, B.; Cremer, T.; Neumann, J. Correlative Microscopy of Individual Cells: Sequential Application of Microscopic Systems with Increasing Resolution to Study the Nuclear Landscape. Methods Mol. Biol. 2013, 1042, 299–336. [Google Scholar]
- Lloyd, D. Oscillations, Synchrony and Deterministic Chaos. Prog. Bot. 2009, 70, 69–91. [Google Scholar]
- Prigogine, I. Time, Structure and Fluctuations. Nobel Lect. Chem. 1977, 263–285. [Google Scholar] [CrossRef] [Green Version]
- Prigogine, I.; Stengers, I. Order out of Chaos: Man’s New Dialogue with Nature; Bantam Books: New York, NY, USA, 1984; ISBN 9780553343632. [Google Scholar]
- Olemskoi, A.I.; Khomenko, A.V.; Olemskoi, D.A. Field theory of self-organization. Phys. A Stat. Mech. Appl. 2004, 332, 185–206. [Google Scholar] [CrossRef]
- Zhukotsky, A.V.; Butusova, N.N.; Shchegolev, A.J.; Kogan, E.M. A vector model of structural regulation in cell nucleus following exposure to Phenobarbital. Biofizika 1985, 177–179. [Google Scholar]
- Gates, R.R.; Ruggles Gates, R. Nucleoli and related nuclear structures. Bot. Rev. 1942, 8, 337–409. [Google Scholar] [CrossRef]
- Maszewski, J.; Kwiatkowska, M. Number, size, and transcriptional activity of nucleoli during different periods of interphase in antheridial filaments of Chara vulgaris L. Folia Histochem. Cytobiol. 1984, 22, 9–19. [Google Scholar]
- Almeira, N.; Risau-Gusman, S. Role of transcriptional bursts in cellular oscillations. J. Biol. 2017, 426, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Fukaya, T.; Lim, B.; Levine, M. Enhancer Control of Transcriptional Bursting. Cell 2016, 166, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Symmons, O.; Raj, A. What’s Luck Got to Do with It: Single Cells, Multiple Fates, and Biological Nondeterminism. Mol. Cell 2016, 62, 788–802. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ni, T.; Wang, W.; Liu, F. Gene transcription in bursting: A unified mode for realizing accuracy and stochasticity. Biol. Rev. 2019, 94, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Budylin, A. Related changes in RNA synthesis and DNA superhelicity during starvation of Ehrlich ascites tumour cells. Proc. Latv. Acad. Sci. 1990, 10, 90–94. [Google Scholar]
- Lloyd, D.; Murray, D.B. The temporal architecture of eukaryotic growth. FEBS Lett. 2006, 580, 2830–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchyia, M.; Wong, S.T.; Yeo, Z.X.; Colosimo, A.; Palumbo, M.C.; Farina, L.; Crescenzi, M.; Mazzola, A.; Negri, R.; Bianchi, M.M.; et al. Gene expression waves. FEBS J. 2007, 274, 2878–2886. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.; Murray, D.B. Ultradian metronome: Timekeeper for orchestration of cellular coherence. Trends Biochem. Sci. 2005, 30, 373–377. [Google Scholar] [CrossRef]
- Liu, S.; Chen, H.; Ronquist, S.; Seaman, L.; Ceglia, N.; Meixner, W.; Chen, P.Y.; Higgins, G.; Baldi, P.; Smale, S.; et al. Genome Architecture Mediates Transcriptional Control of Human Myogenic Reprogramming. iScience 2018, 6, 232–246. [Google Scholar] [CrossRef] [Green Version]
- Pederson, T.; King, M.C.; Marko, J.F. Forces, fluctuations, and self-organization in the nucleus. Mol. Biol. Cell 2015, 26, 3915–3919. [Google Scholar] [CrossRef] [Green Version]
- Vickaryous, M.K.; Hall, B.K. Human cell type diversity, evolution, development, and classification with special reference to cells derived from the neural crest. Biol. Rev. 2006, 81, 425–455. [Google Scholar] [CrossRef]
- Cope, F.O.; Willie, J.J. (Eds.) Carcinogenesis and Apoptosis: Paradigms and paradoxes in cell cycle and differentiation. In Apoptosis: The Molecular Basis of Cell Death; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1991; pp. 61–77. [Google Scholar]
- Lorenz, E.N. Deterministic Nonperiodic Flow. J. Atmos. Sci. 1963, 20, 130–141. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Roach, H.I. Epigenetic selection as a possible component of transdifferentiation. Further study of the commitment of hypertrophic chondrocytes to become osteocytes. Mech. Ageing Dev. 1996, 87, 165–182. [Google Scholar] [CrossRef]
- Noble, R.; Noble, D. Harnessing stochasticity: How do organisms make choices? Chaos 2018, 28, 106309. [Google Scholar] [CrossRef] [Green Version]
- Wapenaar, K.; Snieder, R. Determinism: Chaos tamed. Nature 2007, 447, 643. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J. “Tamed” chaos in embryonal development and carcinogenesis: A holistic view. Proc. Latv. Acad. Sci. Sect. B 2000, ½, 1–8. [Google Scholar]
- Mojtahedi, M.; Skupin, A.; Zhou, J.; Castaño, I.G.; Leong-Quong, R.Y.Y.; Chang, H.; Trachana, K.; Giuliani, A.; Huang, S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016, 14, e2000640. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.; Hemberg, M.; Barahona, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef]
- Erenpreiss, J.O. Current Concepts of Malignant Growth; Zinâtne Publish: Riga, Lative, 1993. [Google Scholar]
- Illmensee, K.; Mintz, B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc. Natl. Acad. Sci. USA 1976, 73, 549–553. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Kleinsmith, L.J.; Pierce, G.B. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964, 24, 1544–1551. [Google Scholar]
- Pierce, G.B.; Barry Pierce, G. Carcinoma is to Embryology as Mutation is to Genetics. Am. Zool. 1985, 25, 707–712. [Google Scholar] [CrossRef]
- Pisco, A.O.; Huang, S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: “What does not kill me strengthens me”. Br. J. Cancer 2015, 112, 1725–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmina, K.; Gerashchenko, B.I.; Hausmann, M.; Vainshelbaum, N.M.; Zayakin, P.; Erenpreiss, J.; Freivalds, T.; Cragg, M.; Erenpreisa, J. When Three Isn’t a Crowd: A Digyny Concept for Treatment-Resistant, Near-Triploid Human Cancers. Genes 2019, 10, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, L.C. Experimental production of testicular teratomas in mice. Proc. Natl. Acad. Sci. USA 1964, 52, 654–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainshelbaum, N.M.; Zayakin, P.; Kleina, R.; Giuliani, A.; Erenpreisa, J. Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes. Genes 2019, 10, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinnitsky, V.B. Oncogerminative hypothesis of tumor formation. Med. Hypotheses 1993, 40, 19–27. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Cai, Z.; Jin, G.; Peng, D.; Pan, B.S.; Zhang, X.; Han, F.; Xu, X.; Lin, H.K. Abnormal gametogenesis induced by p53 deficiency promotes tumor progression and drug resistance. Cell Discov. 2018, 4, 54. [Google Scholar] [CrossRef]
- Kastan, M.B. Wild-Type p53: Tumors Can’t Stand It. Cell 2007, 128, 837–840. [Google Scholar] [CrossRef] [Green Version]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evan, G.; Harrington, E.; Fanidi, A.; Land, H.; Amati, B.; Bennett, M. Integrated control of cell proliferation and cell death by the c-myc oncogene. Philos. Trans. R. Soc. Lond. B 1994, 345, 269–275. [Google Scholar]
- Tyler, A.L.; Crawford, D.C.; Pendergrass, S.A. Detecting and characterizing pleiotropy: New methods for uncovering the connection between the complexity of genomic architecture and multiple phenotypes- session introduction. Biocomputing 2014, 183. [Google Scholar]
- Baryshev, M.; Inashkina, I.; Salmina, K.; Huna, A.; Jackson, T.R.; Erenpreisa, J. DNA methylation of the Oct4A enhancers in embryonal carcinoma cells after etoposide treatment is associated with alternative splicing and altered pluripotency in reversibly senescent cells. Cell Cycle 2018, 17, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Salmiņa, K.; Belyayev, A.; Inashkina, I.; Cragg, M.S. Survival at the Brink. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Academic Press: Cambridge, MA, USA, 2017; pp. 275–294. [Google Scholar]
- Kossiakoff, A.; Sweet, W.N.; Seymour, S.J.; Biemer, S.M. Systems Engineering Principles and Practice; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Tun, K.; Menghini, M.; D’Andrea, L.; Dhar, P.; Tanaka, H.; Giuliani, A. Why so Few Drug Targets: A Mathematical Explanation? Curr. Comput. 2011, 7, 206–213. [Google Scholar] [CrossRef]
- Illidge, T. Polyploid giant cells provide a survival mechanism for p53 mutant cells after dna damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conant, G.C. Rapid reorganization of the transcriptional regulatory network after genome duplication in yeast. Proc. Biol. Sci. 2010, 277, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosieniak, G.; Sikora, E. Polyploidy: The link between senescence and cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Cragg, M.S. Accelerated Senescence of Cancer Stem Cells: A Failure to Thrive or a Route to Survival? In Senescence Physiology or Pathology; Dorszewska, J., Kozubsk, W., Eds.; InTech: Rijeka, Croatia, 2017; pp. 45–62. [Google Scholar]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M. Cancer: A de-repression of a default survival program common to all cells? BioEssays 2012, 34, 72–82. [Google Scholar] [CrossRef]
- Walther, V.; Hiley, C.T.; Shibata, D.; Swanton, C.; Turner, P.E.; Maley, C.C. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat. Rev. Clin. Oncol. 2015, 12, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350. [Google Scholar] [PubMed]
- Vinogradov, A.E. Human transcriptome nexuses: Basic-eukaryotic and metazoan. Genomics 2010, 95, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystem 2019, 181, 82–87. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef]
- Kondrashov, A.S. Evolutionary genetics of life cycles. Annu. Rev. Ecol. Syst. 1997, 28, 391–435. [Google Scholar] [CrossRef]
- Raikov, I.B. The Protozoon Nucleus–Morphology and Evolution; Springer: Berlin, Germany, 1982. [Google Scholar]
- Demin, S.Y.; Berdieva, M.A.; Goodkov, A.V. Cyclic Polyploidy in Obligate Agamic Amoebae. Cell Tissue Biol. 2019, 13, 242–246. [Google Scholar] [CrossRef]
- Bell, G. The origin and early evolution of germ cells as illustrated by the Volvocales. In The Origin and Evolution of Sex; Allan R Liss, Inc.: New York, NY, USA, 1985; pp. 221–256. [Google Scholar]
- Berdieva, M.; Demin, S.; Goodkov, A. Amoeba proteus and ploidy cycles: From simple model to complex issues. Protistology 2019, 3, 166–173. [Google Scholar] [CrossRef]
- Maciver, S.K. Asexual Amoebae Escape Muller’s Ratchet through Polyploidy. Trends Parasitol. 2016, 32, 855–862. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Klimovich, A.; Anokhin, B.; Anton-Erxleben, F.; Hamm, M.J.; Lange, C.; Bosch, T.C.G. Naturally occurring tumours in the basal metazoan Hydra. Nat. Commun. 2014, 5, 4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjundiah, W. Cellular slime mold development as a paradigm for the transition from Unicellular to Multicellular life. In Multicellularity. Origins and Evolution; Kal, N., Stuart, N., Eds.; The MIT Press: Cambridge, UK, 2016; pp. 105–130. [Google Scholar]
- Ledbetter, D.H. Chaos in the embryo. Nat. Med. 2009, 15, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Peaston, A.E.; Knowles, B.B.; Hutchison, K.W. Genome plasticity in the mouse oocyte and early embryo. Biochem. Soc. Trans. 2007, 35, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Zernicka-Goetz, M.; Huang, S. Stochasticity versus determinism in development: A false dichotomy? Nat. Rev. Genet. 2010, 11, 743–744. [Google Scholar] [CrossRef] [PubMed]
- Lotem, J.; Sachs, L. Epigenetics wins over genetics: Induction of differentiation in tumor cells. Semin. Cancer Biol. 2002, 12, 339–346. [Google Scholar] [CrossRef]
- Erenpreisa, J. Janis Olgerts Erenpreiss and his school of cancer research: Commemorating the 90th anniversary. Proc. Latv. Acad. Sci. Sect. B 2019, 73, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B.; Mallm, J.-P.; Sharma, T.; Mark Welch, D.B.; Rippe, K.; Eils, R.; Langowski, J.; Olins, A.L.; Olins, D.E. Nucleosome repositioning during differentiation of a human myeloid leukemia cell line. Nucleus 2017, 8, 188–204. [Google Scholar] [CrossRef] [Green Version]
- Amson, R.; Karp, J.E.; Telerman, A. Lessons from tumor reversion for cancer treatment. Curr. Opin. Oncol. 2013, 25, 59–65. [Google Scholar] [CrossRef]
- Lipkin, G. Plasticity of the cancer cell: Implications for epigenetic control of melanoma and other malignancies. J. Investig. Dermayol. 2008, 128, 2152–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, S. Differentiation of malignant to benign cells. J. Theor. Biol. 1971, 31, 429–451. [Google Scholar] [CrossRef]
- Huang, S. Back to the biology in systems biology: What can we learn from biomolecular networks? Brief. Funct. Genom. Proteom. 2004, 2, 279–297. [Google Scholar] [CrossRef]
- Li, Q.; Wennborg, A.; Aurell, E.; Dekel, E.; Zou, J.-Z.; Xu, Y.; Huang, S.; Ernberg, I. Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc. Natl. Acad. Sci. USA 2016, 113, 2672–2677. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.C.X.; Tonelli, F.M.P.; Vieira, A.L.G.; Kihara, A.H.; Ulrich, H.; Resende, R.R. Studying complex system: Calcium oscillations as attractor of cell differentiation. Integr. Biol. 2016, 8, 130–148. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Strickland, S.; Mahdavi, V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978, 15, 393–403. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Agrawal, I.; Gong, Z. Reversion of tumor hepatocytes to normal hepatocytes during liver tumor regression in an oncogene-expressing transgenic zebrafish model. Dis. Models Mech. 2019, 12, dmm039578. [Google Scholar] [CrossRef] [Green Version]
- Ishay-Ronen, D.; Christofori, G. Targeting Cancer Cell Metastasis by Converting Cancer Cells into Fat. Cancer Res. 2019, 79, 5471–5475. [Google Scholar] [CrossRef]
- Weaver, W. Science and complexity. Am. Sci. 1948, 36, 536–544. [Google Scholar] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erenpreisa, J.; Giuliani, A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int. J. Mol. Sci. 2020, 21, 240. https://doi.org/10.3390/ijms21010240
Erenpreisa J, Giuliani A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. International Journal of Molecular Sciences. 2020; 21(1):240. https://doi.org/10.3390/ijms21010240
Chicago/Turabian StyleErenpreisa, Jekaterina, and Alessandro Giuliani. 2020. "Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics" International Journal of Molecular Sciences 21, no. 1: 240. https://doi.org/10.3390/ijms21010240
APA StyleErenpreisa, J., & Giuliani, A. (2020). Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. International Journal of Molecular Sciences, 21(1), 240. https://doi.org/10.3390/ijms21010240